
Синдром Криглера-Найяра – причины, симптомы, диагностика и лечение
Синдром Криглера-Найяра – генетическое заболевание из класса ферментопатий, характеризующееся нарушением одного из звеньев процесса обезвреживания и выведения билирубина – конъюгации. Симптомами этого состояния являются желтуха печеночного генеза и тяжелые неврологические нарушения, которые могут привести к летальному исходу еще в младенческом возрасте. Диагностика синдрома Криглера-Найяра производится посредством биохимических проб и определения уровня неконъюгированного билирубина в плазме крови, а также молекулярно-генетическими методиками. Специфического лечения заболевания не существует (за исключением трансплантации печени), терапия сводится к увеличению разрушения и элиминации билирубина из организма (гемосорбция, фототерапия, плазмаферез, прием барбитуратов).
Общие сведения
Синдром Криглера-Найяра – тяжелое генетическое заболевание, характеризующееся нарушением связывания билирубина с глюкуроновой кислотой, что является ключевым этапом его обезвреживания и выведения из организма. Впервые это заболевание было описано в 1952 году двумя американскими педиатрами – Джоном Криглером и Виктором Найяром. Дальнейшее изучение синдрома Криглера-Найяра показало, что это состояние имеет генетическую природу и аутосомно-рецессивный характер наследования, кроме того, удалось выявить две клинические разновидности данной патологии. Заболевание достаточно редкое, поэтому точные цифры встречаемости не определены – большинство исследователей полагает, что она находится на уровне 1:1 000 000. Половое распределение больных синдромом Криглера-Найяра не имеет особенностей, заболевание с одинаковой частотой поражает как мальчиков, так и девочек. В лечении этого состояния крайне важна ранняя (в идеале – пренатальная) диагностика, так как от своевременности начатой терапии очень сильно зависят прогноз заболевания и качество жизни больного.
Синдром Криглера-Найяра
Причины синдрома Криглера-Найяра
Синдром Криглера-Найяра относят к классу ферментопатий (по другой классификации – к группе неконъюгированных гипербилирубинемий), причина этого заболевания кроется в недостаточности уридиндифосфатглюкуронидазы 1, функцией которой является связывание билирубина с двумя молекулами глюкуроновой кислоты. В итоге этого биохимического процесса билирубин становится способным растворяться в воде, выводиться в составе желчи и, главное, значительно падает его токсичность. При синдроме Криглера-Найяра этот процесс резко замедлен или не происходит совсем, вследствие чего возникает задержка элиминации билирубина из организма и его накопление. Билирубин обладает выраженной нейротоксичностью, при повышении концентрации в крови это вещество начинает откладываться в тканях кожных покровов и слизистых оболочек, приводя к развитию желтухи. Когда концентрация билирубина превышает определенный порог, соединение начинает проникать через гематоэнцефалический барьер в головной мозг, приводя к тяжелой энцефалопатии (особенно повреждаются базальные ядра). При отсутствии лечения больные синдромом Криглера-Найяра погибают от многочисленных неврологических расстройств и нарастающей печеночной комы.
Причиной низкой активности уридиндифосфатглюкуронидазы являются мутации гена UGT1A1, который располагается на 2-й хромосоме, отвечает за аминокислотную последовательность и выделение этого фермента. Помимо синдрома Криглера-Найяра дефекты этого гена могут приводить к ряду других нарушений билирубинового обмена наследственного характера – синдрому Жильбера, транзиторной неонатальной билирубинемии семейного типа. Механизм наследования мутаций гена UGT1A1 при синдроме Криглера-Найяра аутосомно-рецессивный. При этом описано несколько вариантов возможного повреждения этого гена, которые приводят к разному течению данного заболевания.
Классификация и симптомы синдрома Криглера-Найяра
В настоящее время описаны две основные клинические формы синдрома Криглера-Найяра, в основном различающиеся между собой тяжестью проявлений и прогнозом заболевания. Это обусловлено типом генетического дефекта в UGT1A1. Первый тип заболевания (СКН-1) вызывается миссенс-мутациями, приводящими к появлению неполноценного фермента, имеющего сигнальную последовательность аминокислот, характерную для подвергающихся внутриклеточной утилизации белков. Таким образом, при этой форме дефект гена поражает кодирующие участки (экзоны), что вызывает развитие патологии у гомозигот. Вскоре после своего образования уридиндифосфатглюкуронидаза 1 разрушается и конъюгации билирубина не происходит совсем.
Синдром Криглера-Найяра 1-го типа характеризуется тяжелым и стремительным течением – первые признаки гипербилирубинемии в виде желтухи обнаруживаются уже через несколько часов после рождения. Со временем к ним присоединяются неврологические нарушения – нистагм, судорожные приступы, иногда возникает опистотонус. Желтуха сохраняется на протяжении всей жизни ребенка, его умственное развитие резко отстает от такового у сверстников, симптомы заболевания неуклонно нарастают даже при интенсивном лечении. Обычно больные синдромом Криглера-Найяра 1-го типа умирают на протяжении первого года жизни из-за интоксикации билирубином и поражения базальных подкорковых ядер (ядерная энцефалопатия).
Причиной синдрома Криглера-Найяра 2-го типа также являются миссенс-мутации гена UGT1A1, однако они могут возникать как в кодирующей последовательности (экзонах), так и в промоторе – участке, отвечающем за экспрессию данного гена. У большинства больных СКН-2 наблюдается наличие на одной хромосоме дефекта экзона, на другой – промотора, то есть, такие лица являются компаунд-гетерозиготами. Результатом нарушения является продукция дефектной формы фермента уридиндифосфатглюкуронидазы, которая не разрушается, но имеет пониженную (на уровне 20-25% от нормы) функциональную активность. Поэтому синдром Криглера-Найяра 2-го типа характеризуется менее тяжелой клинической картиной и более благоприятным прогнозом.
В первые месяцы и даже годы жизни больных синдром Криглера-Найяра этого типа нередко проявляется только незначительной желтухой, при отсутствии лечения к подростковому периоду могут развиваться неврологические отклонения. В ряде случаев, особенно при правильно назначенных терапевтических мероприятиях, никаких нарушений со стороны центральной нервной системы не возникает вовсе. Проявления желтухи различной степени выраженности у больных синдромом Криглера-Найяра 2-го типа могут сохраняться на протяжении всей жизни и нередко расцениваются как индикатор осложнений и ухудшения состояния пациента. С возрастом иногда появляется нистагм, могут регистрироваться судорожные припадки, однако течение и выраженность симптомов заболевания всецело зависят от качества лечения и выполнения рекомендаций специалистов.
Диагностика синдрома Криглера-Найяра
Диагностика синдрома Криглера-Найяра производится на основании данных общего осмотра ребенка, биохимических исследований крови, желчи и мочи, молекулярно-генетических анализов. При осмотре выявляется желтуха, возникшая в первые часы (при СКН-1) или месяцы (СКН-2) жизни, признаки неврологических нарушений (опистотонус, нистагм, длительное сохранение транзиторных рефлексов). У больных 2-м типом синдрома Криглера-Найяра неврологические расстройства могут регистрироваться во взрослом возрасте, тогда как у детей наблюдается только желтуха. Также с возрастом могут присоединяться такие проявления, как нейросенсорная глухота или хореоатетоз.
При биохимическом исследовании крови выявляется выраженная непрямая гипербилирубинемия (вплоть 200-350 мкмоль/л), отсутствие (при синдроме Криглера-Найяра 1-го типа) или резкое снижение концентрации прямого билирубина. Конъюгированная фракция этого соединения отсутствует в желчи при СКН-1 и присутствует в незначительных количествах при СКН-2. Фенобарбиталовая проба при синдроме Криглера-Найяра положительна только в случае наличия уридиндифосфатглюкуронидазы, то есть при СКН-2. Изучение концентрации неконъюгированного билирубина в моче показывает его увеличение. Молекулярно-генетическая диагностика синдрома Криглера-Найяра производится врачом-генетиком – он совершает прямое секвенирование последовательности гена UGT1A1 с целью выявления мутаций. При отягощенной по этому заболеванию наследственности у родителей может осуществляться пренатальная диагностика патологии. Дифференциальный диагноз следует проводить с обычной транзиторной желтухой новорожденных и синдромом Жильбера.
Лечение синдрома Криглера-Найяра
Специфического или этиотропного лечения синдрома Криглера-Найяра на сегодняшний день не существует, все терапевтические мероприятия назначаются для ускорения распада билирубина, его выведения из организма и защиты ЦНС. Особых отличий в терапии 1-го или 2-го типа заболевания нет (за исключением активизации микросомального окисления барбитуратами, которая не производится при 1-м типе), однако при СКН-1 лечение лишь незначительно оттягивает наступление летального исхода. Самым радикальным методом лечения синдрома Криглера-Найяра в настоящее время является операция по аллотрансплантации печени от родственника или генетически сходного донора – в этом органе происходит образование уридиндифосфатглюкуронидазы.
Синдром Криглера-Найяра 2-го типа лечат назначением умеренных доз барбитуратов для активации окисления билирубина и увеличения образования нужного фермента. Кроме того, показаны плазмаферез, гемосорбция, заместительное переливание крови – все эти процедуры направлены на удаление неконъюгированного билирубина из организма. Неплохие результаты у больных синдромом Криглера-Найяра дает фототерапия – облучение кожных покровов приводит к частичному разрушению билирубина и освобождению рецепторов тканей для новых порций этого токсина, что снижает его концентрацию в крови. Правильный питьевой режим и повышенное потребление жидкости ускоряет выведение токсина из организма, поэтому следует избегать обезвоживания. Необходим постоянный мониторинг уровня этого вещества в плазме крови, особенно опасным считается его количество свыше 300-340 мкмоль/л – при такой концентрации билирубин становится способным проникать через гематоэнцефалический барьер.
Прогноз и профилактика синдрома Криглера-Найяра
Прогноз синдрома Криглера-Найяра 1-го типа исключительно плохой – из-за полного отсутствия активности фермента уридиндифосфатглюкуронидазы 1 больные умирают на протяжении первого года жизни из-за осложнений ядерной энцефалопатии. Течение СКН-2 зависит от таких факторов, как выраженность проявлений, своевременность диагностики и начала лечения, соблюдения рекомендаций специалистов, наличия или отсутствия сопутствующих заболеваний. В большинстве случаев прогноз относительно благоприятный – больные синдромом Криглера-Найяра 2-го типа могут прожить до преклонного возраста, из характерных проявлений патологии их может беспокоить только желтуха. Профилактика этого состояния возможна только в рамках консультации генетика для родителей, имеющих отягощенную наследственность по этому заболеванию, а также при помощи пренатальной диагностики.
www.krasotaimedicina.ru
Синдром Криглера Найяра
Синдром Криглера-Найяра возникает из-за недостатка или дефицита фермента уридиндифосфата глюкуронозилтрансферазы. Описаны формы 1 и 2 типа заболевания.
Это редкое наследственное нарушение метаболизма билирубина. Билирубин, химическое вещество, образованное в печени вследствие распада крови, не может быть разрушено при этом заболевании.
Избыточный билирубин приводит к желтухе. Болезнь типа 1 является тяжелой, вскоре после рождения. Тип 2 (также называемый синдромом Ариаса) появляется позже, обычно в позднем младенчестве или детстве.
Дети с заболеванием типа 1 имеют очень высокий уровень билирубина. Умирают от накопления желчных пигментов в головном мозге до достижения возраста одного года, хотя возможна выживаемость до взрослой жизни. Лечение включает фототерапию и трансплантацию печени.

Повреждение мозга менее вероятно при синдроме Криглера-Найяра 2-го типа. Пострадавшие дети обычно имеют менее выраженную гипербилирубинемию (повышенные уровни билирубина), выживают до взрослой жизни. Болезнь реагирует на лечение фенобарбиталом.
Причины, факторы риска, заболеваемость
Синдром Криглера-Наджара возникает из-за недостатка или дефицита фермента уридиндифосфата глюкуронозилтрансферазы.
Это фермент печени, необходимый для изменения билирубина в форму, которую можно удалить из организма через желчь. При его отсутствии или дефиците билирубин не может быть разрушен и накапливается в организме.
Это приводит к желтухе, желтому обесцвечиванию кожи, глаз. Избыточный билирубин повреждает мозг, мышцы, нервы. Мутации гена UGT1A1 отвечают за синдром.
При Криглер-Наджаре типа 1 практически отсутствует функциональная активность фермента. Пациенты с типом 2 имеют до 10% от нормальной активности фермента.
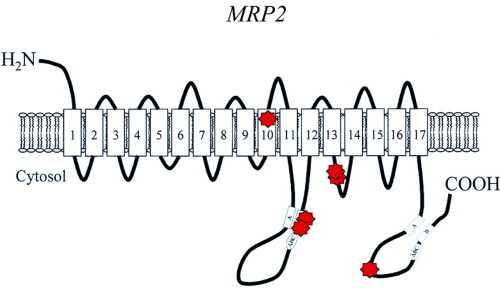
Синдром является унаследованным. Он передается аутосомно-рецессивным способом. Для выявления серьезного заболевания ребенку требуется копия дефектного гена обоих родителей. Родители, у которых есть только один дефектный ген, называются носителями. Они имеют половину ферментативной активности нормального человека.
Синдром Найяра чрезвычайно редок, всего несколько сотен случаев, описаны в мировой литературе. Происходит у менее 1 на 1 000 000 новорожденных. Поражает в равной степени все расы. Брак между кровными родственниками является фактором риска.
Признаки, симптомы
- Тип 1 характеризуется интенсивной желтухой в первые дни жизни с желтым обесцвечиванием кожи, глаз. Эта желтуха, сохраняется после. Накопление билирубина (особенно нерастворимой в воде формы, называемой неконъюгированным билирубином) в головном мозге, наносит ему ущерб, что приводит к изменениям мышления.
За этим может последовать тяжелая форма повреждения головного мозга, называемая kernicterus (билирубиновая энцефалопатия). Симптомы kernicterus включают низкий мышечный тонус, глухоту, летаргию. Состояние часто является фатальным.
До появления фототерапии большинство пациентов с синдромом Криглера-Найяра типа 1 умирало в младенчестве или раннем детстве.
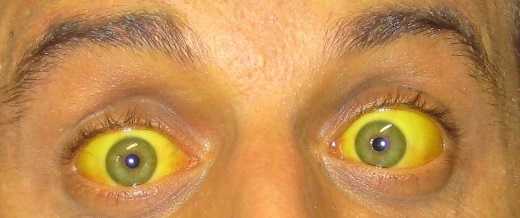
- Тип 2 менее выражен, появляется позже, обычно в позднем младенчестве или детстве. Хотя необычный, kernicterus может возникать во всех возрастах. Пациенты с заболеванием типа 2 редко могут вырабатывать нужные вещества во время таких состояний, как голодание, интеркуррентная болезнь.
Диагностика
Диагностика основана на тестах, которые оценивают функцию печени. К ним относятся:
- Измерение конъюгированного (связанного) и неконъюгированного (несвязанного) билирубина крови. Также измеряется общий уровень билирубина.
- Биопсия печени.
- Анализ ферментов, при котором оценивается уровень фермента уридиндифосфата глюкуронозилтрансферазы.
Клиническое подозрение вызывает развитие желтухи в первый день рождения (патологическая желтуха), которая сохраняется после.
Лечение
Фототерапия превращает билирубин в растворимые формы, которые можно выводить с мочой. Болезнь типа 2 менее выражена и поддается лечению с помощью фенобарбитала.
Тип 1
До появления фототерапии большинство пациентов с синдромом Криглера-Наджара типа 1 умирало в раннем детстве.
Фототерапия или светотерапия
Метод, при котором пациент подвергается воздействию определенных длин волн света. Фототерапия превращает билирубин в растворимые формы, которые выводятся с мочой. Фототерапию иногда дополняют методикой, называемой плазменным обменом. При этой процедуре избыточный билирубин из крови отфильтровывается.
Ранняя трансплантация печени (до наступления повреждения головного мозга) может спасти жизни.
- Соединения кальция иногда используются для связывания и удаления билирубина. Химические вещества, такие как олово протопорфирин или олово-мезопорфирин помогают уменьшить уровень билирубина в качестве чрезвычайной ситуации, но эффект является временным.
Синдром типа 2
Тип 2 менее выражен, поддается лечению с помощью фенобарбитала. Этот препарат относится к группе, называемой барбитуратами. Однократная доза фенобарбитала поддерживает клинически безопасные концентрации билирубина в плазме.
Прогноз
Тип 1 тяжелый и может привести к смерти в детстве, если его не лечить. Младенцы, которые выживают, нуждаются в ежедневном лечении, поскольку желтуха имеет тенденцию сохраняться.
Ожидаемая продолжительность жизни для этого типа составляет 30 лет. Ущерб головного мозга определяется в большинстве случаев.
Болезнь типа 2 менее выражена, имеет меньше симптомов и меньше повреждений органов.
Осложнения
Криглер Найяр может вызвать повреждение головного мозга (kernicterus) и хроническую желтуху.
Понравилась статья? Поделись с друзьями:
Загрузка…ovp1.ru
Синдром Криглера-Найяра
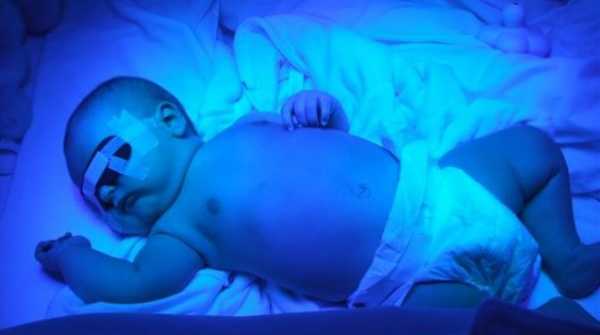
ФототерапияФототерапия – лечение светом. В основе фототерапии лежит физическое свойство импульсов высокоинтенсивного света с определенной длиной волны проникать в структуру кожи.
Фототерапия проводится пациентам с синдромами Криглера-Найяра обоих типов. Этот метод лечения позволяет уменьшить токсическое влияние билирубина и ускорить процесс его элиминации из организма. Фототерапия способствует переводу билирубина в растворимые формы, что позволяет экскретировать его с мочой.
Лечебное действие фототерапии обеспечивается преимущественно за счет синего света. Наиболее часто для фототерапии используются люминесцентные лампы синего света. Могут применяться комбинации: 4 лампы синего света и 2 лампы дневного света.
Максимальный перерыв между сеансами фототерапии (без ущерба для эффективности) составляет не более 2-4 часов. Наиболее оптимальным вариантом проведения фототерапии для большинства новорожденных с синдромом Криглера-Найяра Iтипа является последовательное чередование сеансов фототерапии с перерывами на кормление. При быстром нарастании уровня билирубина фототерапия может проводиться в непрерывном режиме.
У пациентов с синдромом Криглера-Найяра I типа сеансы фототерапии следует сочетать с введениями плазмы крови. Фототерапию у пациентов с синдромом Криглера-Найяра I типа следует рассматривать как подготовку к трансплантации печени.
У пациентов с синдромом Криглера-Найяра II типа эффективность фототерапии снижается через 3-4 года применения, что связано с уменьшением соотношения площади поверхности кожи к массе тела пациента.
При проведении фототерапии, с целью профилактики дегидратации, важно поддерживать баланс жидкости в организме. Редким осложнением фототерапии является развитие синдрома «бронзового ребенка», который может возникать у детей с холестатической желтухой и проявляется коричневой пигментацией кожи, слизистых оболочек и мочи. После прекращения фототерапии эти нарушения исчезают.
www.smed.ru
Синдром Криглера-Наджара (врожденная негемолитическая неконъюгированная билирубинемия)
Что такое Синдром Криглера-Наджара (врожденная негемолитическая неконъюгированная билирубинемия) –
Заболевание является тяжелой формой нарушения обмена билирубина за счет нарушения процесса глюкуронирования вследствие врожденного дефицита фермента глюкуронилтрансферазы.
Что провоцирует / Причины Синдрома Криглера-Наджара (врожденной негемолитической неконъюгированной билирубинемии):
Синдром Криглера-Наджара возникает у лиц с врожденной недостаточностью билирубин-гликозилтрансферазы. Вследствие этого нарушается соединение билирубина с глюкуроновой кислотой и в крови накапливается большое количество неконъюгированного (свободного) билирубина. Дефицит этого фермента приводит к тому, что нарушается преобразование в гепатоците таких субстратов, как салицилаты, кортикостероиды, ментол и др. В крови акапливается большое количество свободного билирубина, являющегося токсичным для организма человека. Новорожденные с таким дефектом не жизнеспособны и вскоре погибают от интоксикации, обусловленной нарушением обмена билирубина. Этот вид гипербилирубинемии следует дифференцировать с физиологической желтухой новорожденных и врожденной гемолитической желтухой – нормобластозом плода. Общим для них является повышение в сыворотке крови свободного билирубина. При физиологической желтухе причина гипербилирубинемии кроется в незрелости конъюгационной системы печени. К 7-10-му дню жизни ребенка эта желтуха проходит. Диагноз нормобластоза плода основывается на выраженной анемии и высоком титре антирезусных антител у матери.
Симптомы Синдрома Криглера-Наджара (врожденной негемолитической неконъюгированной билирубинемии):
Особенности клинических проявлений:
Синдром имеет две генетически гетерогенные формы.
Заболевание передается по аутосомно-рецессивному типу (I тип). Характерна интенсивная желтуха с повышением уровня непрямого билирубина. Фенобарбитал концентрацию билирубина не снижает, появляются симптомы поражения ЦНС. Энцефалопатия при этом является ведущей в клиническом течении заболевания и часто приводит к летальному исходу в течение нескольких недель или месяцев Поражение ядер головного мозга чрезвычайно редко. Гепатоциты, по-видимому, не способны присоединять вторую молекулу глюкуроновой кислоты к моноглюкурониду билирубина. Голодание и интеркуррентные инфекции часто ведут к новому подъему уровня билирубина сыворотки крови.
Заболевание передается по аутосомно-доминантному типу и сопровождается более слабой желтухой (II тип). В кале выявляют значительное количество стеркобилина Уменьшение концентрации билирубина сыворотки достигается применением фенобарбитала. Могут наблюдаться неврологические нарушения.
Диагностика Синдрома Криглера-Наджара (врожденной негемолитической неконъюгированной билирубинемии):
Особенности диагностики:
Лабораторная диагностика: выраженное повышение билирубина в сыворотке, преимущественно неконъюгиро-ванного; снижено содержание билирубина в желчи; активность глюко-ронилтрансферазы в печеночной ткани. При биопсии печени желчные тромбы, перипортальный фиброз.
Лечение Синдрома Криглера-Наджара (врожденной негемолитической неконъюгированной билирубинемии):
Специфического лечения не разработано.
Прогноз:
При I типе синдрома причиной смерти у детей на первом году жизни чаще всего является ядерная желтуха. При II типе больные могут жить многие десятилетия.
К каким докторам следует обращаться если у Вас Синдром Криглера-Наджара (врожденная негемолитическая неконъюгированная билирубинемия):
Гастроэнтеролог
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Синдрома Криглера-Наджара (врожденной негемолитической неконъюгированной билирубинемии), ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Eurolab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом. Клиника Eurolab открыта для Вас круглосуточно.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.
Если Вами ранее были выполнены какие-либо исследования, обязательно возьмите их результаты на консультацию к врачу. Если исследования выполнены не были, мы сделаем все необходимое в нашей клинике или у наших коллег в других клиниках.
У Вас ? Необходимо очень тщательно подходить к состоянию Вашего здоровья в целом. Люди уделяют недостаточно внимания симптомам заболеваний и не осознают, что эти болезни могут быть жизненно опасными. Есть много болезней, которые по началу никак не проявляют себя в нашем организме, но в итоге оказывается, что, к сожалению, их уже лечить слишком поздно. Каждое заболевание имеет свои определенные признаки, характерные внешние проявления – так называемые
Если Вы хотите задать вопрос врачу – воспользуйтесь разделом онлайн консультации, возможно Вы найдете там ответы на свои вопросы и прочитаете советы по уходу за собой. Если Вас интересуют отзывы о клиниках и врачах – попробуйте найти нужную Вам информацию в разделе Вся медицина. Также зарегистрируйтесь на медицинском портале Eurolab, чтобы быть постоянно в курсе последних новостей и обновлений информации на сайте, которые будут автоматически высылаться Вам на почту.
Другие заболевания из группы Болезни желудочно-кишечного тракта:
Если Вас интересуют еще какие-нибудь виды болезней и группы заболеваний человека или у Вас есть какие-либо другие вопросы и предложения – напишите нам, мы обязательно постараемся Вам помочь. www.eurolab.uaСиндром Криглера-Найара – ДНК-диагностика – Центр Молекулярной Генетики
Синдром Криглера-Найяра – наследственная злокачественная неконъюгированная гипербилирубинемия. Тип наследования – аутосомно-рецессивный. Существует 2 типа заболевания, различающиеся по тяжести клинических проявлений.
Синдром Криглера-Найяра I типа (СК-Н I) – самая тяжелая форма заболевания из группы наследственных неконьюгированных гипербилирубинемий, обусловлен мутациями в кодирующей последовательности гена UGT1A1, что приводит к образованию неполноценного фермента уридиндифосфатглюкуронидазы 1, который разрушается. Для СК-Н I типа характерно полное отсутствие фермента УДФ-ГТ1 в гепатоцитах, в связи с чем реакции глюкуронизации билирубина не происходит и непрямой билирубин накапливается в организме, в том числе в ядрах серого вещества, обуславливая тяжелую клинику заболевания. В связи с отсутствием УДФ-ГТ1, фенобарбитал не имеет точки приложения и лечение данным препаратом неэффективно.
Данное заболевание характеризуется злокачественным прогрессирующим течением. Манифестация наступает в первые часы жизни. Клинические проявления заболевания: желтушность склер и кожных покровов, судороги, опистотонус, нистагм, атетоз, замедление умственного развития (билирубиновая энцефалопатия), на ЭЭГ регистрируется медленная активность в задних долях и параксизмальная активность. Биохимические показатели: уровень билирубина в крови выше 200 мкмоль/л. Гистологических изменений в печени не обнаруживают. В желчи полностью отсутствует коньюгированный билирубин. В отсутствии лечебных мероприятий больные погибают в течение первого года жизни от ядерной желтухи.
Синдром Криглера Найяра II (СК-Н II) типа занимает промежуточное положение по тяжести клинических проявления между cиндромом Криглера Найяра I типа и синдромом Жильбера. Заболевание также обусловлено мутациями в кодирующей последовательности гена UGT1A1. Больные с СК-Н II часто являются компаундгетерозиготами, имеющими в одной хромосоме инсерцию в промоторе, а в другой – миссенс-мутацию в экзоне. Кроме того, описаны пациенты, несущие инсерции в промоторной области гена UGT1A1 в гомозиготном состоянии в сочетании со структурной мутацией в экзоне. В основе патогенеза СК-Н II – функциональная недостаточность фермента УДФ-ГТ1.
Манифестация заболевания наступает несколько позже, чем при I типе – от первых месяцев до первых лет. Как правило, больные доживают до юношества без каких-либо неврологических дефектов, хотя билирубиновая энцефалопатия может развиваться в более взрослом возрасте. Клинические проявления заболевания сходны с I типом, но менее тяжелые. Биохимические показатели: уровень билирубина крови менее 200 мкмоль/л. Гистологических изменений в печени не обнаруживают. Желчь пигментирована и содержит билирубин-глюкуронид. Проба с фенобарбиталом положительная
В Центре Молекулярной Генетики проводится прямая ДНК-диагностика данных синдромов, основанная на поиске мутаций во всех экзонах гена UGT1A1, анализе промоторной области.
При проведении пренатальной (дородовой) ДНК-диагностики в отношении конкретного заболевания, имеет смысл на уже имеющемся плодном материале провести диагностику частых анеуплоидий (синдромы Дауна, Эдвардса, Шерешевского-Тернера и др), пункт 54.1. Актуальность данного исследования обусловлена высокой суммарной частотой анеуплоидий – около 1 на 300 новорожденных, и отсутствием необходимости повторного забора плодного материала.
Публикации по теме раздела
Криглера-Найяра синдром
www.dnalab.ru
Синдром Криглера-Наджара

Синдром Криглера-Наджара относится к врождённым пигментным гепатозам, для которых характерно нарушение обмена билирубина, в результате чего имеет место негемолитическая билирубинемия преимущественно за счёт неконъюгированной фракции.
История изучения
Впервые нарушение обмена билирубина, связанное с дефицитом фермента, способствующего присоединению к нему глюкуроновой кислоты, было описано в середине прошлого столетия Наджаром и Криглером, педиатрами из Америки.
Они представили описание шести клинических случаев, которые имели место в трёх семьях, близких друг другу генетически. В разное время специалисты наблюдали тяжёлую желтуху у грудных детей и новорожденных.
Пять из шести наблюдаемых детей не дожили до полутора лет вследствие развития ядерной желтухи. Только один ребёнок справился с данным врождённым дефектом, его организм сумел не допустить интоксикации нервной системы. К 1962 году описано было уже более двадцати детей с данной патологией.
Эпидемиология
На сегодняшний день наиболее вероятным типом наследования считается аутосомно-рецессивный. Среди детей с данной патологией отмечается небольшое преобладание представителей мужского пола.
Этиология и патогенез
Вследствие генетического дефекта, передающегося по наследству, формируется дефицит фермента билирубин-гликозилтрансферазы, который катализирует процесс присоединения глюкуроновой кислоты к молекуле билирубина. Это определяет повышенное накопление в крови ребёнка свободного билирубина.
Кроме того, недостаточное содержание фермента билирубин-гликозилтрансферазы определяет нарушение метаболизма ментола, кортикостероидов и салицилатов, которые в норме преобразуются в клетках печени. Поскольку билирубин является токсическим для организма человека веществом, нарушение его обмена в первые дни и недели жизни ребёнка становится причиной его гибели.
Клиническая картина
В клинической практике различают два типа синдрома Криглера-Наджара. При первом типе патологии билирубин-гликозилтрансфераза в клетках печени не определяется. При втором типе её содержание чрезвычайно низкое.
При первом типе синдрома Криглера-Наджара желтуха развивается в первые дни жизни ребёнка, с каждым днём патологические симптомы нарастают вплоть до развития ядерной желтухи (появляется рвота, гипертермия, анорексия, судорожное подергивание мышц, нарушения работы глазодвигательных мышц, постепенно развиваются психические нарушения), которая не поддаётся лечению возрастными дозами фенобарбитала. При этом отмечается выраженный ответ на применение фототерапии.
При втором типе синдрома Криглера-Наджара развитие ядерной желтухи возможно только в периоде новорожденности, при этом на фоне приёма возрастных доз фенобарбитала отмечается заметное уменьшение явлений желтухи и уровня билирубина в крови. Отмена препарата сопровождается ухудшением общего состояния ребёнка.
Объективно у ребёнка наблюдается желтушность кожных покровов и иктеричность склер. Селезёнка и печень не увеличены в размерах. Стул не обесцвечивается, его окраска практически не меняется. Моча окрашена в золотисто-жёлтый цвет.
Диагностика
При биохимическом исследовании крови определяется повышенное содержание билирубина за счёт неконъюгированной фракции. В желчи содержание билирубина снижено.
Подтвердить диагноз можно при помощи определения уровня активности билирубин-гликозилтрансферазы в гепатоцитах. В биоптате печени выявляют перипортальный фиброз, жёлчные тромбы.
Лечение
Первый тип синдрома является показанием к назначению фототерапии. При втором типе синдрома эффективен приём фенобарбитала. И при первом, и при втором типе синдрома Криглера-Наджара рекомендуется использовать препараты, способствующие снижению циркуляции билирубина в печени и кишечнике. К таким препаратам относятся холестирамин, энтеросорбенты, агар-агар.
Активно разрабатывается методика коррекции дефектного гена и трансплантации печени для лечения данной патологии.
Вы можете посмотреть комментарии или написать свой.
kotikit.ru
описание, причины, симптомы и лечение
Довольно редко можно услышать о таком заболевании, как синдром Криглера-Найяра. Но, к сожалению, этот диагноз могут поставить одному ребенку на миллион. Может показаться, что это крайне редко встречающееся заболевание, но сегодня, в век генетики, мутации выявляются очень часто. Давайте рассмотрим, что это за заболевание и каково лечение в этом случае.
История открытия
Нужно сказать, что открыли этот синдром довольно недавно, в 1952 году прошлого столетия. Два педиатра, Криглер и Найяр, наблюдая за новорожденными детьми, впервые описали необычные симптомы желтухи. Дальнейшие исследования привели к выявлению патологии в работе печени. У деток был сильно повышен непрямой билирубин, который впоследствии оказывал токсическое воздействие на организм в целом. По данным лабораторных исследований, билирубин был повышен до 765 мкмоль/л, при этом он оставался в этих пределах в течение всей жизни ребенка.
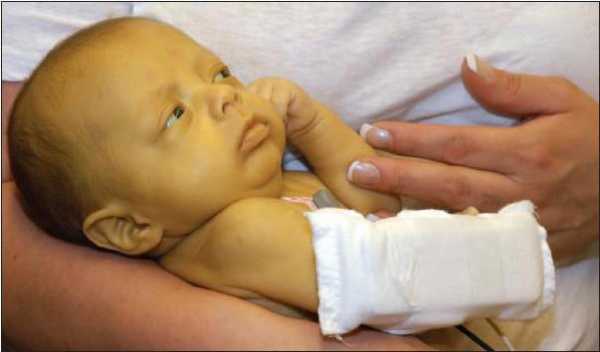 Через некоторое время коллеги выявили подобные симптомы у детей более старшего возраста, но с одной особенностью. Билирубин был повышен всего в 15 раз от нормы и в течение жизни снижался до нормы. Токсического воздействия на организм не было. В результате таких наблюдений заболевание получило свое нынешнее название: синдром Криглера-Найяра, описание которого впервые сделали два педиатра.
Через некоторое время коллеги выявили подобные симптомы у детей более старшего возраста, но с одной особенностью. Билирубин был повышен всего в 15 раз от нормы и в течение жизни снижался до нормы. Токсического воздействия на организм не было. В результате таких наблюдений заболевание получило свое нынешнее название: синдром Криглера-Найяра, описание которого впервые сделали два педиатра.Описание болезни
Синдром Криглера-Найяра является генетическим заболеванием. Клиника болезни выражается в яркой желтухе и тяжелых неврологических нарушениях. Желтуха обнаруживается в первые часы после появления на свет и сохраняется на протяжении всей жизни. Поражения встречаются в одинаковой мере как у мальчиков, так и у девочек. Так как желтушность – это проявление проблем с печенью, то у некоторых пациентов этот орган увеличен в размерах.
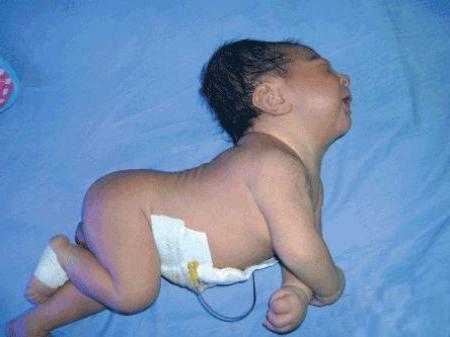 Признаки поражения ЦНС возникают в младенчестве, временами в первые дни жизни. Они выражаются в мышечной напряженности, непроизвольном подергивании глаз, выгибании спины, а также судорогах. Больные дети, как правило, отстают в психическом и физическом развитии. Выявляются два вида этого расстройства. Синдром Криглера-Найяра 1 и 2 типа может иметь различные симптомы.
Признаки поражения ЦНС возникают в младенчестве, временами в первые дни жизни. Они выражаются в мышечной напряженности, непроизвольном подергивании глаз, выгибании спины, а также судорогах. Больные дети, как правило, отстают в психическом и физическом развитии. Выявляются два вида этого расстройства. Синдром Криглера-Найяра 1 и 2 типа может иметь различные симптомы.Симптомы 1 типа
К сожалению, синдром Криглера-Найяра 1 типа характеризуется прогрессирующим течением. Первые симптомы появляются в первые часы жизни. У малыша становится более выраженная желтушность белков глаз и кожных покровов, чем и отличается от обычной послеродовой желтухи. Она не проходит через несколько дней, а к симптомам добавляются судороги, непроизвольные движения тела и глаз. Через время можно заметить замедление умственного развития, связанное с билирубиновой энцефалопатией.
 Показатели свободного билирубина при анализах повышаются до 324—528 мкмоль/л, по сути, это выше нормы в 15-50 раз. Интоксикация головного мозга в этом случае ведет к летальному исходу в течение короткого времени. В исключительных случаях такие дети доживают до школьного возраста.
Показатели свободного билирубина при анализах повышаются до 324—528 мкмоль/л, по сути, это выше нормы в 15-50 раз. Интоксикация головного мозга в этом случае ведет к летальному исходу в течение короткого времени. В исключительных случаях такие дети доживают до школьного возраста.Симптомы 2 типа
Первые признаки болезни появляются значительно позже, чем при 1 типе. Болезнь может проявить себя в первые годы жизни. У некоторых детей желтуха не проявляется вплоть до подросткового возраста, и неврологические отклонения бывают редко. Симптомы сходны с 1 типом, но не настолько тяжелые. Билирубиновая энцефалопатия может возникнуть после перенесенной инфекции или сильного стресса.
 Биохимические показатели крови при 2 типе гораздо ниже – уровень билирубина около 200 мкмоль/л. Этот показатель говорит о том, что активность фермента глюкуронилтрансферазы меньше 20% от нормы. В желчи имеется билирубин-глюгуронид. Диагностика с “Фенобарбиталом” позитивная.
Биохимические показатели крови при 2 типе гораздо ниже – уровень билирубина около 200 мкмоль/л. Этот показатель говорит о том, что активность фермента глюкуронилтрансферазы меньше 20% от нормы. В желчи имеется билирубин-глюгуронид. Диагностика с “Фенобарбиталом” позитивная.Билирубиновая энцефалопатия
Чем же страшен синдром Криглера-Найяра? Симптомы болезни проявляются в отравлении мозга четырьмя фазами. В первой фазе малыш ведет себя апатично и очень вяло. Это проявляется в плохом сосании, расслабленном состоянии, резкой реакции на посторонние звуки. Крик малыша при этом монотонный, он часто срыгивает и может даже открыться рвота, взгляд его блуждающий, как будто он что-то потерял. Дыхание может быть замедленным.
Вторая фаза может длиться от нескольких дней до нескольких месяцев. Ребенок становится напряженным, мышцы тела принимают неестественное положение, ручки постоянно сжаты в кулак, спина выгибается дугой. Крик из монотонного переходит в очень резкий, пропадает сосательный рефлекс и реакция на звуки. Появляются судороги, храп, потеря сознания.
Третья фаза проявляется периодом ложного улучшения состояния. Все предыдущие симптомы на время исчезают.
Четвертая фаза может появиться на 5 месяце жизни и проявляться явными симптомами физической и умственной отсталости. Малыш не держит голову, не следит за движущимися предметами, не реагирует на голос близких людей. У него развиваются судороги, парезы, параличи. К сожалению, отравление мозга при 1 типе происходит очень быстро, и ребенок умирает в младенческом возрасте.
Причины заболевания
Основная причина болезни кроется в генах. В них нарушается процесс образования определенного фермента, который отвечает за выработку билирубина. По большей части этим заболеванием страдает азиатское население планеты. Мутирующий ген передается по аутосомно-рецессивному типу. В этом случае оба родителя малыша могут быть носителями мутации, но сами быть здоровыми. Также может быть носителем один из родителей, тогда вероятность проявления болезни будет 50 на 50%.
 Наследственная мутация гена приводит к тому, что организм не способен связывать свободный билирубин с глюкуроновой кислотой. А это, в свою очередь, ведет к тому, что свободный билирубин отравляет организм, проникая через гематоэнцефалический барьер, который у новорожденных детей не функционирует. Отравлению подвергается мозг ребенка, где накапливается токсический билирубин.
Наследственная мутация гена приводит к тому, что организм не способен связывать свободный билирубин с глюкуроновой кислотой. А это, в свою очередь, ведет к тому, что свободный билирубин отравляет организм, проникая через гематоэнцефалический барьер, который у новорожденных детей не функционирует. Отравлению подвергается мозг ребенка, где накапливается токсический билирубин.Лечение
Для детей, которым поставлен диагноз синдром Криглера-Найяра, лечение направлено на удаление из организма свободного билирубина. Также важно предотвратить развитие токсического повреждения головного мозга.
Для его лечения используются лекарства, повышающие активность уридиндифосфат-глюкуронидазы, фермента, приводящего к необратимым процессам в печени. Для этого применяется “Фенобарбитал” в дозировке до 5 мг на килограмм веса в сутки. Нужно заметить, что он положительно влияет исключительно на синдром Криглера-Найяра 2 типа. При 1 типе организм на “Фенобарбитал” практически не реагирует.
 При обоих типах заболевания проводятся сеансы фототерапии, вводится плазма, выполняются обменные переливания крови. Все процедуры подготавливают к тому, чтобы выполнить трансплантацию печени – для детей с 1 типом это единственный шанс выжить.
При обоих типах заболевания проводятся сеансы фототерапии, вводится плазма, выполняются обменные переливания крови. Все процедуры подготавливают к тому, чтобы выполнить трансплантацию печени – для детей с 1 типом это единственный шанс выжить.Диагностика
На сегодняшний день медицина способна установить причины таких заболеваний, как синдром Криглера-Найяра. Симптомы и методы лечения описаны достаточно давно, а сейчас при помощи ДНК-тестов можно предопределить генетическую предрасположенность к заболеваниям еще внутриутробно. После рождения малыша ДНК-диагностика дает точный ответ о том, есть ли мутация в определенных генах.
 Также при развивающейся желтухе делается проба с “Фенобарбиталом”. Результат анализа показывает тип заболевания.
Также при развивающейся желтухе делается проба с “Фенобарбиталом”. Результат анализа показывает тип заболевания.При подозрении на синдром Криглера-Найяра у родителей собирается анамнез и проводятся ДНК-тесты для подтверждения диагноза.
Профилактика заболевания
Профилактические меры при синдроме Криглера-Найяра состоят в предотвращении появления осложнений.
При синдроме I типа очень важно предотвратить развитие билирубиновой энцефалопатии, потому что именно она приводит к преждевременной гибели больного.
При синдроме II типа профилактика сводится к информированию больного об обстоятельствах, которые могут спровоцировать обострение заболевания. Это осложняющие инфекции, перенапряжение, беременность, принятие спиртного и лекарственных препаратов без контроля лечащего врача. Все это может вызвать повышение билирубина в крови и привести к сильной интоксикации. В этой статье невозможно описать все случаи, ведь синдром Криглера-Найяра (лечение, причины, симптомы которого рассмотрены нами) может проявляться у детей индивидуально.
fb.ru