
причины, симптомы, диагностика и лечение
Фенилкетонурия – наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина. Ранними признаками фенилкетонурии служат рвота, вялость или гиперактивность, запах плесени от мочи и кожи, задержка психомоторного развития; типичные поздние признаки включают олигофрению, отставание в физическом развитии, судороги, экзематозные изменения кожи и др. Скрининг новорожденных на фенилкетонурию проводится еще в родильном доме; последующая диагностика включает молекулярно-генетическое тестирование, определение концентрации фенилаланина в крови, биохимический анализ мочи, ЭЭГ, МРТ головного мозга. Лечение фенилкетонурии заключается в соблюдении специальной диеты.
Общие сведения
Фенилкетонурия (болезнь Феллинга, фенилпировиноградная олигофрения) – врожденная, генетически обусловленная патология, характеризующаяся нарушением гидроксилирования фенилаланина, накоплением аминокислоты и ее метаболитов в физиологических жидкостях и тканях с последующим тяжелым поражением ЦНС. Фенилкетонурия впервые описана А. Феллингом в 1934 г.; встречается с частотой 1 случай на 10 000 новорожденных. В неонатальном периоде фенилкетонурия не имеет клинических проявлений, однако поступление фенилаланина с пищей вызывает манифестацию заболевания уже в первом полугодии жизни, а в дальнейшем приводит к тяжелым нарушениям развития ребенка. Именно поэтому пресимптоматическое выявление фенилкетонурии у новорожденных является важнейшей задачей неонатологии, педиатрии и генетики.
Фенилкетонурия
Причины фенилкетонурии
Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена.
Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу и расположенного на длинном плече 12 хромосомы (локус12q22-q24.1). Это, так называемая, классическая фенилкетонурия I типа, составляющая 98% всех случаев заболевания. Гиперфенилаланинемия может достигать 30 мг% и выше. При отсутствии лечения данный вариант фенилкетонурии сопровождается глубокой умственной отсталостью.
Кроме классической формы, различают атипичные варианты фенилкетонурии, протекающие с той же клинической симптоматикой, но не поддающиеся коррекции диетотерапией. К ним относятся фенилкетонурия II типа (недостаточность дегидроптеринредуктазы), фенилкетонурия III типа (дефицит тетрагидробиоптерина) и другие, более редкие варианты.
Вероятность рождения ребенка, больного фенилкетонурией, повышается при заключении близкородственных браков.
Патогенез фенилкетонурии
В основе классической формы фенилкетонурии лежит недостаточность фермента фенилаланин-4-гидроксилазы, участвующего в конверсии фенилаланина в тирозин в митохондриях гепатоцитов. В свою очередь, производный тирозина – тирамин является исходным продуктом для синтеза катехоламинов (адреналина и норадреналина), а дийодтирозин – для образования тироксина. Кроме этого, результатом метаболизма фенилаланина служит образование пигмента меланина.
Наследственная недостаточность фермента фенилалаиин-4-гидроксилазы при фенилкетонурии приводит к нарушению окисления фенилаланина, поступающего с пищей, в результате чего его концентрация в крови (фенилаланинемия) и спинномозговой жидкости значительно возрастает, а уровень тирозина соответственно падает. Избыточное содержание фенилаланина устраняется путем повышенной экскреции с мочой его метаболитов – фенилпировиноградной, фенилмолочной и фенилуксусной кислот.
Нарушение обмена аминокислот сопровождается нарушением миелинизации нервных волокон, снижением образования нейромедиаторов (дофамина, серотонина и др.), запускающими патогенетические механизмы задержки умственного развития и прогредиентное слабоумие.
Симптомы фенилкетонурии
Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев. С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда – беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома. Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота, которая нередко ошибочно расценивается как проявление пилоростеноза.
Ко второму полугодию становится заметным отставание ребенка в психомоторном развитии. Ребенок становится менее активным, безучастным, перестает узнавать близких, не пытается садиться и вставать на ножки. Аномальный состав мочи и пота обусловливают характерный «мышиный» запах (запах плесени), исходящий от тела. Часто наблюдается шелушение кожи, дерматиты, экзема, склеродермия.
У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия, прогнатия, позднее (после 1,5 лет) прорезывание зубов, гипоплазия эмали. Отмечается задержка речевого развития, а к 3-4 годам выявляется глубокая олигофрения (идиотия) и практически полное отсутствие речи.
Дети с фенилкетонурией имеют диспластическое телосложение, нередко – врожденные пороки сердца, вегетативные дисфункции (потливость, акроцианоз, артериальную гипотонию), страдают запорами. К фенотипическим особенностям детей, страдающих фенилкетонурией, следует отнести светлую кожу, глаза и волосы. Для ребенка с фенилкетонурией характерны специфическая поза «портного» (согнутые в суставах верхние и нижние конечности), тремор рук, шаткая, семенящая походка, гиперкинезы.
Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом, сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет.
При фенилкетонури III типа развивается триада признаков: микроцефалия, олигофрения, спастический тетрапарез.
Диагностика фенилкетонурии
В настоящее время диагностика фенилкетонурии (а также галактоземии, врожденного гипотиреоза, адрено-генитального синдрома и муковисцидоза) входит в программу неонатального скрининга, осуществляемого всем новорожденным.
Скрининг-тест проводится на 3-5 день жизни доношенного и 7 день жизни недоношенного ребенка путем забора образца капиллярной крови на специальный бумажный бланк. При обнаружении гиперфенилаланемии более 2,2 мг% ребенка направляют к детскому генетику для повторного обследования.
Для подтверждения диагноза фенилкетонурии проверяется концентрация фенилаланина и тирозина в крови, определяют активность печеночных ферментов (фенилаланингидроксилазы), выполняется биохимическое исследование мочи (определение кетоновых кислот), метаболитов катехоламинов в моче и др. Дополнительно проводится ЭЭГ и МРТ головного мозга, осмотр ребенка детским неврологом.
Генетический дефект при фенилкетонурии может быть обнаружен еще на этапе беременности в ходе инвазивной пренатальной диагностики плода (хорионбиопсии, амниоцентеза, кордоцентеза).
Дифференциальный диагноз фенилкетонурии проводят с внутричерепной родовой травмой новорожденных, внутриутробными инфекциями, другими нарушениями обмена аминокислот.
Лечение фенилкетонурии
Основополагающим фактором в лечении фенилкетонурии является соблюдение диеты, ограничивающей поступление белка в организм. Лечение рекомендуется начинать при концентрации фенилаланина >6 мг%. Для грудных детей разработаны специальные смеси – Афенилак, Лофенилак; для детей старше 1 года – Тетрафен, Фенил-фри; старше 8 лет – Максамум-ХР и др. Основу диеты составляют низкобелковые продукты – фрукты, овощи, соки, белковые гидролизаты и аминокислотные смеси. Расширение диеты возможно после 18 лет в связи с возрастанием толерантности к фенилаланину. В соответствии с российским законодательством обеспечение лиц, страдающих фенилкетонурией, лечебным питанием, должна осуществляться бесплатно.
Больным назначается прием минеральных соединений, витаминов группы В и др.; по показаниям – ноотропные средства, антиконвульсанты. В комплексной терапии фенилкетонурии широко используется общий массаж, ЛФК, иглорефлексотерапия.
Дети, страдающие фенилкетонурией, находятся под наблюдением участкового педиатра и психоневролога; нередко нуждаются в помощи логопеда и дефектолога. Необходим тщательный мониторинг нервно-психического статуса детей, контроль уровня фенилаланина в крови и показателей электроэнцефалограммы.
Атипичные формы фенилкетонурии, не поддающиеся лечению диетой, требуют назначения гепатопротекторов, противосудорожных средств, заместительной терапии леводопой, 5-гидрокситриптофаном.
Прогноз и профилактика фенилкетонурии
Проведения массового скрининга на фенилкетонурию в неонатальном периоде позволяет организовать раннюю диетотерапию и предотвратить тяжелые церебральные повреждения, нарушения функции печени. При раннем назначении элиминационной диеты при классической фенилкетонурии прогноз развития детей хороший. При поздно начатом лечении прогноз в отношении умственного развития неблагоприятный.
Профилактика осложнений фенилкетонурии заключается в проведении массового скрининга новорожденных, раннего назначения и длительного соблюдения диетического питания.
С целью оценки риска рождения ребенка с фенилкетонурией предварительное генетическое консультирование должны пройти супружеские пары, уже имеющие больного ребенка, состоящие в кровнородственном браке, имеющие родственников с данным заболеванием. Женщины с фенилкетонурией, планирующие беременность, должны соблюдать строгую диету до зачатия и во время беременности для исключения повышения уровня фенилаланина и его метаболитов и нарушения развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
www.krasotaimedicina.ru
симптомы, лечение, признаки у детей и новорожденных, диагностика
Фенилкетонурия – это сложное генетическое заболевание, в основе которого лежит нарушение обмена аминокислот. В результате в организме не происходит трансформации жизненно важной аминокислоты фенилаланина в тирозин.
Эта болезнь достаточно распространена, а выявляется она в среднем в одном случае на 8 тысяч новорожденных. Поскольку организм при таком генетическом нарушении не получает тирозина, это обуславливает множество патологий. Ведь это вещество воздействует на работу гипофиза, щитовидки и надпочечников, а также препятствует откладыванию жира в организме и уменьшает аппетит.
Впервые фенилкетонурия у детей была выявлена в 30-е годы прошлого века ученым А. Феллингом. Первые симптомы заболевания проявляются практически сразу же после рождения, а дальнейшее его прогрессирование может привести к тяжелым осложнениям, в том числе поражению головного мозга, нарушениям развития у ребенка психики и движения, а также токсическому повреждению нервной системы.
Причины фенилкетонурии

Причины фенилкетонурии обусловлены наследственной предрасположенностью. Однако заболевание передается только в том случае, когда оба родителя являются носителями измененного гена.
Этот ген встречается только у двух процентов населения земного шара, при этом человек может быть абсолютно здоров. Однако если мужчина и женщина, являющиеся его носителями, вступают в брак, то они могут передать его своему ребенку. Даже в этом случае вероятность рождения малыша с генетическими нарушениями составляет только 25 процентов.
Также фенилкетонурия может быть обусловлена близкородственным браком, поскольку в этом случае родители могут являться носителями мутированного гена, который кодирует фермент фенилаланин-4-гидроксилаз. Фенилаланин в организме больного человека выводится очень медленно, поэтому при его постоянном поступлении он поражает нервную систему.
Если заболевание было выявлено своевременно, то качество и продолжительность жизни такого ребенка ничем не отличается от других людей. Однако при некачественной диагностике и отсутствии терапии она значительно сокращается.
Симптомы фенилкетонурии

Симптомы фенилкетонурии появляются у ребенка сразу же после рождения. Среди них можно выделить:
- Усталость и вялость;
- Беспокойство;
- Отсутствие интереса к окружающему миру;
- Аллергический дерматит и другие кожные заболевания;
- Судороги;
- Повышенное беспокойство и раздражительность;
- Срыгивания после еды;
- Бледность кожных покровов;
- Нарушение тонуса мышц;
- Специфический запах мочи.
В старшем возрасте симптомы фенилкетонурии проявляются в виде задержки психического и физического развития, а при проведении обследования врачи могут обнаружить микроцефалию.
К тому же больные имеют специфические внешние особенности. У них светлая фарфоровая кожа, белые волосы и голубые глаза. Если болезнь выявлена в первые дни после рождения, то при правильно назначенном лечении и диете ребенок может сохранить шансы на здоровую жизнь.
Новорожденный с этой патологией рождается абсолютно здоровым, а ухудшение состояния наступает впоследствии, поскольку организму не хватает вещества тирозина. Поэтому при раннем выявлении заболевания головной мозг не поражается, а ребенок растет аналогично своим сверстникам.
В том случае, когда фенилкетонурия после рождения не была установлена, а ребенок длительное время употреблял продукты с большим содержанием фенилаланина, наступают необратимые последствия. При его накоплении в жидкостях организма и тканях происходит токсическое отравление нервной системы и головного мозга, а в тяжелых случаях может развиться идиотия.
В возрасте после двух лет у ребенка могут проявиться следующие симптомы:
- Недержание мочи;
- Эпилептические судороги и спазмы;
- Дрожание рук;
- Уменьшение размеров черепа;
- Скованные движения, обусловленные повышенным тонусом мышц;
- Деформирование раковин ушей;
- Нарушения поведения.
При отсутствии диеты в лечении больного возможно развитие психических отклонений, которые становятся причиной инвалидности.
Заболевание может иметь разную симптоматику в зависимости от формы. Всего выделяют три типа, среди которых наиболее распространенной является первый. Он поддается лечению благодаря ограничениям в диете, а болезнь второго и третьего типа может привести к летальному исходу.
Диагностика фенилкетонурии
Фенилкетонурия обычно выявляется на пятый день после рождения. Для этого всем новорожденным проводят перед выпиской обследование, после кормления берут анализ крови. Ее наносят на специальные тестовые полоски, после чего осуществляется анализ, который позволяет выявить эту патологию.
Для подтверждения диагноза проводится анализ крови и мочи, в котором будет заметно повышение уровня фенилаланина. Для предотвращения симптомов заболевания ребенку назначается незамедлительное лечение и специальная диета с ограничением белковых продуктов, которая позволяет ему впоследствии вести нормальную и здоровую жизнь.
Эта генетическая болезнь также может быть диагностирована в процессе визуального осмотра и сбора анамнеза. Однако этот метод использовался еще несколько десятилетий назад, сегодня самыми проверенными являются способы лабораторной молекулярно-генетической диагностики.
Лечение фенилкетонурии

Лечение фенилкетонурии проводится пожизненно, однако наиболее важным периодом является детский возраст до 18 лет. В это время наиболее активно развивается нервная система, а накопление фенилаланина в организме может привести к ее токсическому поражению.
Однако только специальная диета позволит человеку вести здоровую и полноценную жизнь вплоть до самой старости. Врачи таким пациентам советуют продолжать соблюдать диету на протяжении всей жизни, чтобы улучшить ее качество.
На первом году жизни ребенок должен питаться специальными смесями с достаточным количеством незаменимых аминокислот, витаминов и микроэлементов. К ним относятся Фенил Фри, ХР Аналог, Афенилак, MIDмил ФКУ.
Назначить определенную молочную смесь врач может только на основании результатов анализов. В дополнении к искусственным молочным смесям мать должна кормить ребенка грудью для укрепления его иммунной системы, однако дозировку молока должен подбирать специалист на основании проведенных анализов и обследований.
Для детей после одного года для питания помимо прикорма могут быть использованы Изифен, ХР Максамейд и ХР Максамум, а также П-АМ. Эти продукты не содержат фенилаланина, поэтому они не вызывают нарушений в работе внутренних органов и прекрасно перевариваются.
Диета для взрослых и детей при фенилкетонурии назначается сразу же после проведения генетического анализа. В этом случае питание должно быть богато растительными белками, витаминами и микроэлементами. К полностью разрешенным продуктам относятся – картошка, сахар, растительные масла, а также зеленый и черный чай.
Среди запрещенных продуктов можно выделить:
- Мясные и рыбные блюда, а также птица;
- Молочные продукты: сметана, йогурт, сыр, молоко и творог;
- Куриные яйца;
- Мука и макаронные изделия;
- Горох и другие бобовые;
- Кукуруза;
- Шоколад и орехи;
- Желатин.
К разрешенным продуктам, которые необходимо употреблять в ограниченном количестве, относятся – хлеб, сливочное масло, овощи и фрукты, рис, мед, а также щербеты. Также важно, чтобы в организм человека поступали продукты, богатые тирозином.
Это вещество необходимо для полноценного функционирования организма, а в больших количествах оно содержится в грибах и других растительных продуктах. Такое лечение фенилкетонурии позволит пациенту прожить долгую полноценную жизнь.
В последние годы специалистами был разработан препарат, содержащий растительную фенилаланингидроксилазу. Он позволяет контролировать содержание фенилаланина в органах и тканях, не давая ему накапливаться в больших количествах.
Народные средства лечения фенилкетонурии

Фенилкетонурия является опасным и сложным генетическим заболеванием, поэтому ее лечение народными средствами не проводится.Однако возможно использование травяных сборов и настоев для улучшения общего состояния здоровья.
Они могут снять раздражительность и беспокойство у новорожденных, а также восполнить нехватку витаминов и микроэлементов, которые связаны с жесткими ограничениями при диете. Больные могут употреблять мяту, ромашку, календулу, мелиссу, боярышник, пустырник. При этом орехи и сухофрукты смогут укрепить иммунную систему. Использование всех лекарственных препаратов и трав необходимо согласовывать с врачом.
Профилактика фенилкетонурии

Фенилкетонурия у грудничков и новорожденных не может быть предупреждена, поскольку эта болезнь обусловлена генетическим нарушением. Однако для предупреждения опасных последствий болезни необходима ее ранняя диагностика и впоследствии тщательная диета.
При выявлении этого заболевания у одного ребенка в семье необходимо проводить перинатальную диагностику заболевания при последующей беременности, а также возможна медико-генетическая консультация.
Понравилась статья? Поделись с друзьями:
Loading …Также по этой теме:
Not foundnarhiler.ru
3 метода диагностики, основные симптомы и 4 важных правила диетотерапии
О болезни
Фенилкетонурия (ФКУ) – генетическое заболевание, которое связано с нарушением обмена аминокислот, главным образом фенилаланина. Поступление в организм малыша обычной белковой пищи приводит к накоплению в тканях токсических продуктов обмена веществ. Эти соединения необратимо влияют на нервную систему и головной мозг ребёнка, вызывают прогрессирующее слабоумие.
В некоторых странах недуг носит имя первооткрывателя и называется болезнью Фёллинга. Иное название, фенилпировиноградная олигофрения, указывает на патогенез заболевания.
Статистика распространения наследственной болезни отличается в различных странах. Так, в России недуг обнаруживается у одного малыша из 5 – 10 тысяч новорождённых (в зависимости от региона страны). Наиболее безопасной страной в отношении риска развития фенилкетонурии считается Финляндия (1:100000). Турция же занимает первое место по возникновению генетического недуга, один из 2600 новорождённых имеет нарушенный обмен фенилаланина.
Среди больных фенилкетонурией девочки встречаются в 2 раза чаще, чем мальчики. Наследование происходит по аутосомно-рециссивному типу, что означает наличие мутантного гена в обеих семьях родителей. Здоровые носители генетического дефекта могут не догадываться о своей особенности и не иметь никаких внешних проявлений. Ситуация усложняется, когда обладатели мутации вступают в брак и собираются завести детей. Риск появления на свет малыша, больного фенилкетонурией, в таком случае составляет 25%. Половина детей, рождённых в данной семье, останутся бессимптомными носителями наследственного дефекта.

Хотя заболевание может привести к грубым, необратимым нарушениям в развитии малыша и значительно ухудшить качество жизни крохи, неприятных последствий можно избежать. Фенилкетонурия – одна из немногих наследственных болезней, которая эффективно поддаётся лечению. При своевременно принятых мерах малыш практически всегда вырастает здоровым.
Историческая справка
Впервые наследственную природу слабоумия заподозрил норвежский биохимик и психиатр Ивар Асбьёрн Фёллинг. Наблюдая за группой умственно отсталых детей, врач заметил общность клинической картины и обнаружил изменение химического состава мочи больных брата и сестры. Добавив к биологической жидкости раствор хлорного железа, доктор выявил изменение окраски мочи, появление оливково-зелёного оттенка. Наличие изменений анализов у близких родственников дало основание заподозрить наследственные причины заболевания.
Этот метод обнаружения фенилкетонурии не потерял своей актуальности и в современном мире. Определение в моче фенилпировиноградной кислоты специалисты называют пробой Фёллинга в честь норвежского врача.

Как развивается болезнь?
Предугадать развитие фенилкетонурии у новорождённого малыша без применения дополнительных исследований невозможно. Кроха ничем не отличается от своих сверстников, выглядит абсолютно здоровым ребёнком. Изменения происходят внутри организма, когда начинается энтеральное вскармливание, в организм поступают белки грудного молока.
Причина патологии кроется в недостатке определённых ферментов, участвующих в превращении незаменимой аминокислоты фенилаланина в тирозин. Таким образом происходит повышение уровня фенилаланина и его производных в крови и спинномозговой жидкости. Продукты обмена веществ оказывают токсическое действие на клетки головного мозга, нарушают жировой обмен и передачу нервных импульсов между нейронами.
В то же время необходимый для нормального функционирования тирозин, не синтезируется в должном количестве. Эта аминокислота является незаменимой и принимает активное участие в образовании гормонов, нейромедиаторов, пигмента меланина.
Различают несколько клинических форм фенилкетонурии, которые отличаются дефицитом определённого фермента в печени. В 98% возникает ФКУ I типа, связанная с дефектом гена в 12-й хромосоме. Недуг проявляется недостатком фенилаланин-4-гидроксилазы и хорошо поддаётся лечению. II и III типы заболевания относятся к крайне редким недугам и характеризуются тяжёлыми проявлениями, неэффективностью диетотерапии в коррекции обмена аминокислот.
Симптомы классической формы фенилкетонурии
Общие проявления
Если неонатальный скрининг не был проведён в родильном доме, заболевание осталось не распознанным и не были организованы необходимые лечебные мероприятия, то ФКУ проявит себя на первом году жизни крохи. Первые признаки отклонений от нормы родители обнаружат у ребёнка в возрасте 2 – 6 месяцев, когда ранее здоровый карапуз становится вялым и апатичным или наоборот, чрезмерно раздражительным. Приём пищи часто заканчивается срыгиванием, а на коже младенца появляются проявления аллергического дерматита.
Характерная внешность
Поскольку нарушение обмена аминокислот приводит к уменьшению образования меланина, в случае развития заболевания кожные покровы, волосы, радужная оболочка глаз теряют пигмент. Малыши с генетическим заболеванием имеют светлые волосы, тонкую бледную кожу, голубые глаза. Указывать на наследственный недуг может характерный запах пота и мочи крохи. Многие врачи характеризуют его как «мышиный», а связан он с выделением продуктов обмена аминокислот с биологическими жидкостями.

Поражение нервной системы
Первые признаки изменений со стороны нервной системы проявляются вялостью, снижением тонуса мышц крохи. Нередко возникает дистония, появляются непроизвольные сокращения мышц, навязчивые движения, судороги. Возможно развитие органических патологий головного мозга – микроцефалии и гидроцефалии.
Практически у 50% больных фенилкетонурией развивается эпилепсия. Иногда характерные приступы являются первым признаком наследственного заболевания. Пароксизмы плохо поддаются лечению обычными противосудорожными препаратами и значительно снижают качество жизни ребёнка.
Отставание в развитии
С течением болезни малыш быстро теряет интерес к окружающему миру, становится апатичным. Физическое развитие ребёнка так же страдает – кроха поздно овладевает необходимыми навыками. Задержка в психическом развитии становится очевидной, когда возникают проблемы в формировании речи, отставание в умственном развитии.

Исследования показали, что грубые изменения интеллектуальной деятельности развиваются уже через год прогрессирования болезни. Кроха необратимо теряет до 50 баллов IQ ежегодно, что впоследствии приводит к развитию дебильности, имбецилии или идиотии. Поэтому сроки начала лечения фенилкетонурии очень важны.
Неврологические проблемы у детей старшего возраста выражаются в гиперактивности, появлении стереотипий, навязчивых движений кистей рук, лица, языка, покачиваний. Иногда расстройства психики проявляются в шизофреноподобных состояниях.
Грубое отставание в развитии приводит к отсутствию навыка ходьбы у 30% больных детей и алалии, недоразвитии речи, у 60% пациентов.
Разновидности патологии
Транзиторная форма гиперфенилаланинемии
Не всегда недуг протекает классически, иногда повышение уровня фенилаланина носит временный характер, а само заболевание не развивается. Непродолжительное увеличение концентрации аминокислоты в крови ребёнка может быть связано с незрелостью ферментных систем малыша, что наиболее характерно для глубоко недоношенных детей. Такие малыши требуют более детального обследования для определения причины заболевания.
Птерин-зависимые варианты фенилкетонурии
Примерно у 2 % малышей, у которых была выявлена фенилкетонурия в родильном доме и назначена соответствующая диета, возникают неврологические проблемы. Эти симптомы могут указывать на развитие у крохи редкой, птерин-зависимой формы ФКУ (тип II, III по классификации). Клинические проявления атипичных форм ФКУ сходно с течением классической формы, но симптомы прогрессируют быстрее, а диетотерапия не оказывает необходимого эффекта.

Огромную роль в развитии птерин-зависимых форм играет дефицит нейромедиаторов, который возникает из-за нарушения обмена аминокислот. Эти вещества необходимы для передачи нервных импульсов между нервными клетками, без которого нормальное функционирование организма невозможно.
Фенилкетонурия II считается более злокачественной, чем III типа. Грубые нарушения обмена аминокислот приводят к массовой гибели нервных клеток. Нередко малыши с прогрессирующей ФКУ II типа гибнут в возрасте 2 – 3 лет.
Материнская фенилкетонурия
Не всегда больные ФКУ дети рождаются от здоровых родителей. Если беременные женщины с наследственной патологией не соблюдают диеты, высока вероятность рождения малыша с синдромом материнской фенилкетонурии. Токсические вещества проникают через плаценту из материнского организма к плоду и нарушают формирование органов и систем ребёнка. На свет появляются дети с множественными врождёнными пороками развития, микроцефалией, умственной и физической отсталостью.
Диагностика
Медико-генетическое консультирование семьи
Будущим родителям нужно быть особенно внимательными, если в семьях кого-либо из них имели место случаи рождения больных фенилкетонурией детей. Нельзя исключить возможность появления на свет ребёнка с ФКУ, даже если недуг проявился у дальнего родственника. Коварство заболевания кроется в бессимптомном носительстве повреждённого гена абсолютно здоровыми людьми.
Всем парам, у которых имеются подозрения на наличие наследственных болезней в роду, стоит обратиться к врачу-генетику ещё во время планирования малыша. В медико-генетическом центре (МГЦ) с помощью современных методов диагностики можно выявить носительство мутантного гена и рассчитать риск рождения ребёнка с ФКУ и другими генетическими болезнями.
Инвазивные методы диагностики (хорионбиопсии, амниоцентеза, кордоцентеза) могут помочь определить недуг до рождения крохи. При этом исследуется генетический материал, полученный от плода. Данные методы являются травматичными и могут повлечь за собой спонтанное прерывание беременности. Поэтому применение их оправдано лишь при доказанном носительстве мутантных генов у родителей и высоком риске возникновения ФКУ у малыша.
Неонатальный скрининг
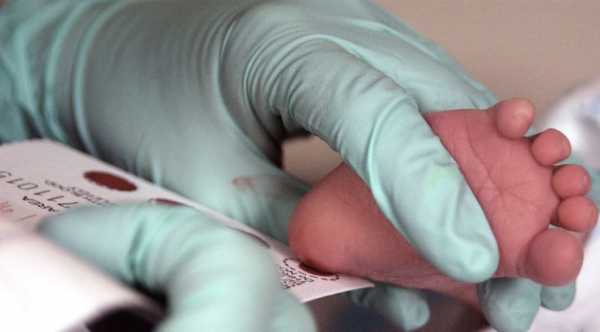
Перед выпиской из родильного дома новорождённых малышей массово обследуют на наследственные заболевания. Для этого важного исследования медработник набирает кровь из пяточки малыша и наносит капельки биологической жидкости на фильтровальную часть тест-бланка. Каждая капля крови предназначена для выявления одного из 5 заболеваний: фенилкетонурии, муковисцидоза, врождённого гипотиреоза, адреногенитального синдрома, галактоземии.
Неонатальный скрининг – очень важное и необходимое всем детям без исключения обследование. Он проводится совершенно бесплатно и направлен на сохранение здоровья нации. Отказываясь от манипуляции, родители совершают огромную ошибку, ведь симптомы наследственных недугов не всегда заметны у новорождённого малыша. Яркие клинические проявления нередко возникают, когда помочь ребёнку уже сложно, а изменения в организме крохи приняли необратимый характер.
Этот метод является достоверным в выявлении фенилкетонурии, если малыш получает достаточное количество энтерального питания, грудного молока. Поэтому новорождённым, которые находятся в отделении реанимации, анализ проводят позже. Отличаются и сроки постановки пробы у доношенных и недоношенных детей. Малышам, которые появились в срок, рекомендован набор капиллярной крови на 4-е сутки жизни. Проведение исследование родившимся преждевременно крохам откладывается до 7-х суток.
Анализ следует проводить натощак, что обеспечит более точный результат и повысит диагностическую значимость теста. Бланк с капельками крови малыша и паспортными данными родителей отправляется в лабораторию медико-генетической консультации, где проводится биохимическое исследование.
Родителям нужно обратить внимание на правильность заполнения бланка, корректность своих контактных данных. На проведение исследования уходит в среднем 10 дней, семья к этому времени обычно находится дома. В случае обнаружения положительного результата теста, родителям нужно будет в кратчайшие сроки обратиться в медико-генетический центр, а неправильные контактные данные задержат дальнейшее обследование малыша.
Обследование ребёнка в МГЦ
Чтобы подтвердить заболевание у младенца и установить его причину, малышу придётся пройти многоэтапную процедуру обследования. Повторные биохимические анализы помогут подтвердить повышение уровня аминокислоты в крови и принять решение о необходимости диетотерапии. Проводится исследование уровней фенилаланина и тирозина, активность печёночных ферментов, определение продуктов обмена аминокислот в моче.
Затем осуществляется молекулярно-генетическая диагностика, которая позволяет выявить мутацию в гене РАН, ответственном за развитие фенилкетонурии. С помощью данных методов можно определить и бессимптомное носительство заболевания.
Лечение ФКУ
Самым эффективным методом терапии классической формы фенилкетонурии считается правильно организованная диетотерапия. Только исключив из рациона крохи продукты, содержащие большое количество аминокислоты, можно ожидать положительных результатов от лечения.
Особенности диетотерапии
Правила подбора пищевых продуктов
Фенилкетонурия у детей – непростое заболевание и правильно подобрать рацион для малыша может только врач. Выбирая разрешённые продукты и рассчитывая их объём, доктор учитывает некоторые индивидуальные особенности:
- какая из форм болезни присутствует у малыша;
- степень тяжести недуга, уровень фенилаланина в крови;
- возраст крохи;
- особенности физического развития непоседы;
- физиологическую потребность растущего организма в белке;
- как переносит малыш поступление небольшого количества фенилаланина с пищей.
Аминокислота фенилаланин является незаменимой, а значит может поступать в организм только с белковой пищей. Растущий ребёнок нуждается в достаточном поступлении нутриентов, ведь около 40% пищевого фениаланина расходуется на выстраивание собственных белков. Получается, что полностью исключить из рациона крохи опасные продукты также невозможно. Поэтому в первые дни диетотерапии лабораторно оценивается, как организм крохи реагирует на небольшое поступление аминокислоты с продуктами питания.
Возможно ли грудное вскармливание при фенилкетонурии?
Поскольку, в большинстве случаев, болезнь определяется ещё в первые недели жизни крохи, становится вопрос о безопасности кормления малыша грудным молоком. На самом деле, грудное вскармливание необходимо для нормально развития ребёнка, становления его иммунной системы. Но применение его оправдано лишь в случаях, когда мать придерживается чётких правил и строго контролирует употребляемый малышом объём съеденной пищи.
Для безопасного кормления лучше использовать свежесцеженное грудное молоко, допустимое количество которого рассчитывает врач исходя из массы тела малыша и его потребности в фенилаланине.
Чем младше ребёнок, тем выше его потребность в аминокислоте. Например, допустимый объём фенилаланина для новорождённого малыша составляет 90 – 60 мг/кг массы тела в сутки, в то время как необходимое количество аминокислоты для школьника снижается до 10 – 20 мг/кг массы тела. Количество фенилаланина в продуктах можно рассчитать, учитывая, что 1 г белка содержит около 50 мг аминокислоты.
Специализированные смеси
Существуют специально разработанные продукты для рационального питания больных фенилкетонурий малышей. Они изготавливаются на основе гидролизатов белка или смеси аминокислот, которые полностью или частично лишены фенилаланина. Питание лечебными смесями направлено на восполнение организма крохи необходимым белком, достаточное поступление питательных веществ. Эти смеси могут использоваться как заменители грудного молока или применяться, как добавка к основному рациону для старших детей.
Состав лечебных смесей для детей разного возраста отличается. Например, для ребят до года подойдут такие продукты, как «Афенилак», «Лофеналак». Старшим детям показаны «Тетрафен», «Фенил-фри», «Максамум – ХР».

«Пищевой светофор»
С каждым годом малыш растёт, и его потребности в продуктах питания увеличиваются. Приходит время вводить прикорм, пробовать новые блюда. Родителям нужно помнить об опасности фенилаланина для мозга малыша, выбирать только разрешённые врачом продукты.
Рацион больного ребёнка значительно отличается от питания сверстников. Прикорм начинается с введения овощей и фруктов, затем следует добавлять безбелковые макароны, некоторые виды круп. Составлять первое меню малыша и контролировать содержание аминокислот в крови на фоне изменения рациона должен лечащий врач.
Все продукты разделяют на группы по содержанию в них фенилаланина:
- красный список.
Рекомендуется полностью исключить продукты этого перечня, поскольку они богаты белками. Это мясо и субпродукты, рыба, яйца, хлеб, гречневая, ячневая крупы, хлопья « Геркулес»;
- жёлтый список.
Допустимо использовать небольшое количество продуктов из этой группы под контролем анализа крови. Возможно введение кукурузной и рисовой крупы, картофеля, молочных продуктов с низким содержанием белка;
- зелёный список.
Эти продукты составляют основу питания больных фенилкетонурией детей старшего возраста: мука кукурузная и рисовая, большинство овощей, фрукты и ягоды, сладости, сливочное масло.
При применении готовых продуктов нужно внимательно читать инструкцию на упаковке. Употребление бананов, сухофруктов, бобовых может повысить концентрацию фенилаланина, поэтому их приём стоит ограничить.

Другие методы терапии
Применение медикаментов оправдано лишь для лечения птерин-зависимых форм ФКУ, когда диета не оказывает надлежащего эффекта. В таких ситуациях используют препараты L-дофы и 5-гидроокситриптофана.
В остальных случаях возможно использование витаминотерапии, минеральных комплексов, как дополнение к диете. При необходимости назначаются ноотропные препараты, антиконвульсанты. Работа с логопедами, психологами, массаж, физиотерапия так же оказывают положительный результат в лечении недуга.
Прогноз и профилактика
Течение классической формы заболевания значительно не влияет на продолжительность жизни человека. Но без надлежащего лечения тяжёлые симптомы недуга значительно сказываются на интеллекте и качестве жизни пациента. В случае рациональной диетотерапии и своевременного контроля уровня фенилаланина в крови, дети с ФКУ не отличаются от своих сверстников и хорошо адаптируются в обществе. Атипичные формы недуга нередко приводят к инвалидизации и смерти ребёнка, поскольку лечение редко оказывает хороший результат.
Специалисты рекомендуют соблюдать диету в течение всей жизни человека, но некоторые послабления возможны по достижению ребёнком 18 лет. Особенно внимательными нужно быть беременным женщинам, больным ФКУ, чтобы избежать синдрома материнской фенилкетонурии у ребёнка.
Профилактика заключается в предупреждении близкородственных браков, медико-генетическом консультировании семей из группы риска по развитию ФКУ, проведении массового обследования новорождённых.

Выводы
Болезнь у малыша – огромный стресс и испытание для молодых родителей, особенно если недуг носит наследственный характер. Но фенилкетонурия – одна из немногих генетических болезней, мероприятия по лечению которой приносят хорошие результаты.
Родителям нужно понимать важность неонатального скрининга и своевременно начатой, правильно подобранной диетотерапии. Стоит внимательно соблюдать все рекомендации лечащего врача и тогда такой страшный диагноз, как фенилкетонурия не станет приговором для ребёнка.
kroha.info
Фенилкетонурия профилактика. Существует ли профилактика фенилкетонурии? Механизм развития заболевания
ФКУ (аббревиатура заболевания фенилкотурия) – это редкий наследственный недуг, причина возникновения которого кроется в расстройстве обмена аминокислот. Организм не в состоянии утилизировать фенилаланин. Он поступает вместе с пищей и накапливается в теле пациента и медленно отравляет его изнутри.
Подобно яду, он действует на нервную систему больного и вызывает соответствующую симптоматику. Вследствие этого, ФКУ имеет и другое название – фенилпировиноградная олигофрения. Это, пожалуй, единственное наследственное заболевание, которое можно целиком купировать. Дети, рожденные с этим диагнозом, при должном лечении вырастают нормальными и здоровыми.
Фенилкетонурия у новорожденных достаточно редко встречается. В разных странах этот показатель отличается. Так, в России 1 случай заболевания приходится на 10 000. Великобритания имеет большую частоту данного показателя – 1 на 5 000. В Африке патология практически не встречается. Согласно статистике, девочки болеют ФКУ чаще, чем мальчики примерно в 2 раза.
Пусковым фактором для развития заболевания является наследственность. ФКУ зарождается в двух случаях:
- При близкородственных браках повышается вероятность проявления дефектных генов, в том числе растет шанс возникновения фенилкетонурии;
- Из-за мутации в области 12 хромосомы ввиду разнообразных причин.
ФКУ возникает спонтанно в, казалось бы, благополучной семье, без известных случаев данного заболевания у родственников. Наличие патологического гена в организме пациента приводит к тому, что печень не вырабатывает специальный фермент фенилаланин – 4 – гидроксилазу. Это и является причиной развития клинических проявлений заболевания.
Патогенез
Фенилкетонурия возникает в тех случаях, когда оба родителя – папа и мама, передали малышу склонность к этой патологии. Они сами могут не болеть, но сочетание у ребенка двух дефектных генов от родителей наделяют его шансом к ФКУ 25%. Вероятность такого совпадения довольно мала, поэтому и феникетонурия встречается нечасто.
Фермент, отсутствующий при болезни, должен превращать поступающую с белковой пищей аминокислоту фенилаланин в тирозин. Последний составляет пигмент меланин, другие ферменты, гормоны, то есть нужен для адекватной работы организма. Дефектный ген у больных ФКУ приводит к тому, что фенилаланин не идет по нормальной цепочке превращений. Он не становится тирозином, а тот не выполняет свои функциональные обязанности.
В итоге у больных аминокислота превращается в соединения, которых в организме не должно быть вовсе – фенилпировиноградная и фенилмолочная кислоты, фенилэтиламин и ортофенилацетат. Эти вещества копятся, словно мусор, и вызывают:
- Нарушение процессов жирового обмена в головном мозге;
- Недостаток нейромедиаторов, передающих нервные импульсы между клетками;
- Интоксикацию, которая отравляет ЦНС.
Это негативное воздействие значительно и необратимо снижает интеллект больного. У малышей быстро возникает олигофрения – умственная отсталость.
Симптомы
Клиника ФКУ имеет довольно яркое и специфическое проявление. Она заметна уже в первый год жизни ребенка. Основными симптомами являются:
- Вялость и незаинтересованность в окружающем мире;
- Повышенная плаксивость;
- Расстройство тонуса мышц;
- Судороги;
- Сниженная пигментация кожных покровов, волос и радужки глаз;
- Склеродермия;
- Эпилептические припадки;
- «Мышиный» запах мочи.
Однако главным признаком является нарушение в психоречевом развитии и микроцефалия. Это не означает, что заболевание проявит себя сразу же. Фенилкетонурия у новорожденных может не давать внешне заметных симптомов. Определить проблему «налицо» становится возможным лишь на 2-6 месяц. Физически больные младенцы меньше отличаются от сверстников, нежели чем в психическом плане. Нередко головка ребенка меньше, чем положено его возрасту.
Зубки у таких деток прорезываются позже, сидеть и ходить они тоже не начинают в срок. Внешность ребенка с ФКУ характерна – это бледная белая кожа и светлые волосы с глазами. Это объясняется нарушением пигментации, вызванной отсутствием тирозина. Такая кожа чувствительна и склонна к появлению высыпаний под воздействием ультрафиолетового излучения.
Дети в возрасте года не могут голосом показывать эмоции и понимать чужую речь. Их мимика невыразительна. С возрастом симптомы увеличиваются, словно снежный ком. Наблюдаются нарушения в поведении таких деток, психическая и умственная отсталость. Без лечения клинические проявления будут лишь прогрессировать.
Диагностика ФКУ
Достоверным способом установления наличия заболевания является медико-генетическая экспертиза. Сегодня она носит характер неонатального скрининга для массового обследования новорожденных. Такая процедура эффективна для выявления самых распространенных наследственных патологий. В это число входит и фенилкетонурия.
К дополнительному обследованию, направленному на точное установление болезни, относится биохимический анализ крови и общий мочи. ФКУ подтверждается в случае:
- Повышенного уровня фенилаланина, больше 900 мкмоль/л;
- Нахождения фенилпировиноградной, фенилмолочной или фенилуксусной кислот;
- Положительной пробы Феллинга.
Лечение
Каким бы опасным ни было заболевание, с ним можно бороться, сохранив нормальное качество жизни. Главной линией в лечении ФКУ является диетотерапия. Помимо этого, внимания заслуживают:
- Энзимотерапия фенилаланингидроксилазой, фенилаланинаммониалиазой;
- Прием тетрагидробиоптерина – «Сапроптерин»;
- Метод больших нейтральных аминокислот.
Эффективно показало себя применение пищевых гликомакропептидов. Они снижают концентрацию фенилаланина в крови больного и его головном мозге. Такая терапия направляется на адекватное физическое развитие детей. Создан экспериментальный метод лечения ФКУ. Его смысл состоит в непосредственном введении гена фенилаланингидроксилазы в поврежденные клетки печени. В России данные методики еще не нашли себе применения.
Прогноз и профилактика
flgso.ru
Чем Опасно Нарушение? Как его Лечить?
Фенилкетонурия — это редкое нарушение, при котором организм ребенка при рождении не способен расщеплять аминокислоту под названием фенилаланин.
Что такое фенилкетонурия?
Фенилкетонурия — это редкое нарушение, при котором организм ребенка при рождении не способен расщеплять аминокислоту под названием фенилаланин.
Каковы симптомы фенилкетонурии?
Вначале симптомы отсутствуют. Однако в течение первых месяцев жизни в зависимости от тяжести заболевания у ребенка могут проявиться определенные симптомы, а именно:
- Маленький размер головы (так называемая микроцефалия).
- Гиперактивность.
- Запах плесени и затхлости от мочи, дыхания или кожи.
- Более светлые кожа, волосы и глаза, чем у братьев и сестер.
- Наличие кожных нарушений, таких как экзема.
- Подрагивание рук или ног.
- Тремор.
- Судороги.
- Замедленное развитие.
- Поведенческие нарушения.
- Психические расстройства.
- Перманентная умственная отсталость.
Как диагностируется фенилкетонурия?
В большинстве стран всех детей при рождении обследуют на предмет целого ряда возможных заболеваний и нарушений. Одной из процедур обследования является сдача анализа крови, который помимо прочего позволяет выявить наличие фенилаланин-гидроксилазы — фермента, необходимого для расщепления фенилаланина. Если этот фермент отсутствует, для подтверждения диагноза необходимы дополнительные анализы мочи и крови.
Можно ли предотвратить развитие фенилкетонурии?
Поскольку фенилкетонурия является генетическим нарушением, передающимся от родителей к детям, предотвратить его невозможно. Единственным способом определения наличия дефектного гена является генетическое тестирование. У носителя гена могут отсутствовать какие бы то ни было симптомы или проявления фенилкетонурии. А для передачи заболевания ребенку у каждого из родителей должен быть соответствующий ген. Кроме того, ряд обследований во время беременности (биопсия ворсин хориона или амниоцентез) позволяют заранее выявить фенилкетонурию.
При наличии этого заболевания необходимо соблюдать жесткую диету (с минимальным количеством фенилаланина) как до беременности, так и во время нее. Отложения фенилаланина наносят ущерб развивающемуся плоду, даже если он не унаследовал дефектный ген.
Как лечится фенилкетонурия?
Наиболее важной в лечении фенилкетонурии является диета, ограничивающая употребление содержащих фенилаланин продуктов. Новорожденным при диагностировании этого заболевания необходимо питаться специальной смесью, которую можно смешивать с небольшим количеством грудного молока или обычной детской смеси. Но баланс этот установить довольно сложно. Дело в том, что ребенку для нормального развития все же необходимо определенное количество фенилаланина. В то же время, слишком большое его количество может нанести ущерб.
В целом, детям с фенилкетонурией необходимо соблюдать диету с низким содержанием фенилаланина. Он содержится в большинстве белковых продуктов, включая мясо, рыбу, молоко, сыр, яйца, орехи, сою и бобы. Также он встречается и в небелковых продуктах, а именно в ряде овощей и фруктов, хлебе и пиве. Подсластитель аспартам, вырабатывающий фенилаланин при переваривании, также следует исключить из рациона.
Управление США по надзору за качеством пищевых продуктов и медикаментов подтвердило эффективность применения сапроптерина дигидрохлорида (Куван) для лечения фенилкетонурии. Куван помогает организму расщеплять фенилаланин. В то же время, препарат подходит не всем, и он все равно не способен справиться с любым количеством этого вещества. Принимающие его люди должны параллельно соблюдать диету.
Последствия фенилкетонурии
Из-за ограничения допустимого рациона довольно сложно обеспечивать организм всеми необходимыми питательными веществами. Именно поэтому для страдающих фенилкетонурией людей разработали специальную питательную смесь. Также им может потребоваться прием пищевых добавок. К примеру, рыбий жир способен заменить некоторые длинноцепочные жирные кислоты (базовые строительные блоки липидов), отсутствующие в рамках диеты. Эти жирные кислоты способствуют неврологическому развитию.
У каждого страдающего фенилкетонурией человека есть определенный уровень фенилаланина, который он способен усвоить. Именно поэтому вам необходимо проконсультироваться со специалистом и разработать индивидуальную диету. А регулярная сдача анализов крови и частое посещение врача помогут держать ситуацию под контролем. Помните, что организму для нормального развития необходимо определенное количество фенилаланина. В то же время, он не должен вредить вашему здоровью.
Так или иначе, диета при фенилкетонурии действительно ограничивает человека во многом. Но ее все равно необходимо соблюдать. Четкое следование диете на протяжении всей жизни позволяет человеку сохранить психическое и физиологическое здоровье. А со стрессом, сопровождающим многочисленные ограничения, поможет эффективно справиться поддержка друзей, родных и психологов.
Вопросы, которые следует задать врачу
- Нужно ли мне пройти генетическое тестирование для проверки на фенилкетонурию?
- Если у меня диагностировано это заболевание, что мне нужно предпринять во время беременности для защиты плода?
- Как часто нужно приводить страдающего фенилкетонурией ребенка на осмотр?
- Что определяет тяжесть фенилкетонурии?
- Чем нужно кормить ребенка?
- От каких продуктов ему нужно отказаться?
- Что делать, если ребенок съел «запрещенный» продукт?
- Если у первого ребенка диагностировали фенилкетонурию, значит ли это, что и следующий мой ребенок также родится с этим заболеванием?
mojdoktor.pro
Фенилкетонурия (ФКУ) у детей – симптомы болезни, профилактика и лечение Фенилкетонурии (ФКУ) у детей, причины заболевания и его диагностика на EUROLAB
Что такое Фенилкетонурия (ФКУ) у детей –
Фенилкетонурия (ФКУ) у детей — генетическая болезнь, которая характеризуется нарушениями обмена фенилаланина и бывает у 1 из 8000–15 000 новорожденных. Форм фенилкетонурии (ФКУ) всего 4, но существует 400 разных мутаций и метаболические фенотипы заболевания.
Фенилкетонурия — наследственная аминоацидопатия, при которой снижается интеллект ребенка, и возникает неврологический дефицит.
Фенилкетонурия I (классическая или тяжелая) – это аутосомно-рецессивное заболевание, которое возникает вследствие мутации гена фенилаланингидроксилазы. В основе заболевания лежит нехватка фенилаланин-4-гидроксилазы которая обеспечивает превращение фенилаланина в тирозин, результатом чего становится накопление фенилаланина и его метаболитов в тканях и физиологических жидкостях организма ребенка.
Отдельную группу представляю атипичные варианты фенилкетонурии. При них симптомы очень похожи на таковые при классическом варианте заболевания. Но нет положительных продвижений по показателям развития ребенка, даже если проводить нужную диетотерапию. Такие варианты объясняются нехваткой дегидроптеринредуктазы, тетрагидроптерина, гуанозин-5-трифосфатциклогидролазы, 6-пирувоилтетрагидроптеринсинтазы и пр.
Фенилкетонурия II (атипичная) — аутосомно-рецессивная болезнь, при которой генный дефект находится в коротком плече хромосомы 4. Характеризуется она нехваткой дегидроптеринредуктазы, что приводит к нарушению восстановления активной формы тетрагидробиоптерина, а в спинномозговой жидкости и сыворотке крови снижается уровень фолатов. Результат таких изменений – метаболические блоки в механизмах превращения фенилаланина в тирозин. Заболевание было выявлено еще в конце 20-го века.
Фенилкетонурия III (атипичная) — аутосомно-рецессивная болезнь, которая вызвана недостаточностью 6-пирувоилтетрагидроптеринсинтазы. Он принимает участие в организме в процессе создания тетрагидробиоптерина из дигидронеоптеринтрифосфата, что было открыто в конце 20-го столетия. Нарушения сходы с таковыми при выше описанной (второй) форме.
Примаптеринурия — атипичная фенилкетонурия у детей с легкой гиперфенилаланинемией, у которых присутствует в больших количества в моче примаптерин и часть его производных, а в спинномозговой жидкости нормальная концентрация нейромедиаторных метаболитов.
Материнская ФКУ – болезнь, при которой снижается уровень интеллекта (вплоть до умственной отсталости) среди потомства женщин, которые больны фенилкетонурией и не сидели на специальной идете, когда были совершеннолетними.
Есть предположения, что при материнской ФКУ нарушения в развитии белого вещества мозга ответственны за формирование неврологического дефицита. Было проведено исследование в 2008 году Кочем и его командой. У младенца, рожденного от матери с ФКУ, при аутопсии головного было найдено некоторое количество патологических изменений: вентикуломегалия, низкий вес мозга, задержка миелинизации (признаков астроцитоза не наблюдалось), гипоплазия белого вещества.
В некоторых странах СНГ применяется условная классификация рассматриваемого заболевания по уровню содержания в сыворотке крови фенилаланина:
|
Название формы |
Уровень фенилаланина |
|
классическая |
выше 20 мг% (1200 мкмоль/л) |
|
средняя |
10,1–20 мг% (600–1200 мкмоль/л) |
|
легкая |
до 8 мг% (480 мкмоль/л) |

Что провоцирует / Причины Фенилкетонурии (ФКУ) у детей:
Основная причина фенилкетонурии у детей – нехватка фермента с названием фенилаланин-4-гидроксилаз. Из-за его отсутствия происходит скопление в жидкостях и тканях фенилаланина в большом количестве, который воздействует токсически на ЦНС ребенка. Результатом становится нарушения метаболизма гормонов, обмена белков, транспорта аминокислот, обмена глико- и липопротеидов.
Причиной ФКУ у детей может стать:
- хронический алкоголизм матери и/или отца
- инфекционные воспалительные процессы половых органов родителей
- влияние на организм отца и матери пагубных факторов и пр.
Патогенез (что происходит?) во время Фенилкетонурии (ФКУ) у детей:
Основа патогенеза фенилкетонурии (ФКУ) у детей является нехватка фермента фенилаланин-4-гидроксилаза, который обеспечивает превращение фенилаланина в тирозин. Производные фенилаланина (фенилпировиноградная, фенилуксусная, фенилмолочная кислоты, фенилэтилламин, фенилацетилглютамин) и само вещество скапливаются в тканях. Следствием является негативное влияние на центральную нервную систему. Возникают разные нарушения, в том числе в обмене катехоламинов и серотонина.
Симптомы Фенилкетонурии (ФКУ) у детей:
Новорожденный ребенок не похож на больного. Симптомы фенилкетонурии (ФКУ) начинают быть заметны в возрасте 2-6 месяцев. Типичные проявления:
- отсутствие интереса к окружающему миру
- выраженная вялость
- рвота
- беспокойство
- повышенная раздражительность
С 6 месяцев у малыше заметно отставание в психическом развитии. У меньшинства детей это олигофрения в слабой степени. А более чем у половины детей фиксируют идитию. Рост малыша с ФКУ может быть нормальным или сниженным. Зубки режутся поздно, череп может иметь размеры меньше нормы. Сидеть и ходить ребенок с фенилкетонурией начинает поздно.
Детей с рассматриваемым диагнозом можно отличить по позе и походке. Они широко расставляют ноги, сгибая их в тазобедренном и коленных суставах. Шаги мелкие. При ходьбе ребенок покачивается. Сидят они в так называемом положении портного – поджав ноги, поскольку у них повышен мышечный тонус.
При фенилкетонурии (ФКУ) дети обычно имеют голубой цвет глаз и светлый оттенок волос. Кожа почти не пигментирована. От ребенка слышен «мышиный» запах. В некоторых случаях у больного могут быть припадки эпилепсии, но они проходят по мере взросления ребенка.
Другие типичные симптомы ФКУ у детей:
- дермографизм
- потливость
- повышенная чувствительность к солнечным лучам и травмам
- акроцианоз
- тяжёлая экзема
- дерматит
- склонность к запорам
- артериальная гипотония
- расстройства аутистического спектра
- гиперактивность
Если не провести вовремя лечение, уровень интеллекта ребенка будет составлять менее 50. В возрасте 18 месяцев могут появиться судорожные приступы. Исчезают они спонтанно. В раннем возрасте приступы часто проходят в форме инфантильных спазмов, далее становятся тоникоклоническими припадками.
Диагностика Фенилкетонурии (ФКУ) у детей:
Для диагностики фенилкетонурии (ФКУ) у детей определяют содержание крови уровней фенилаланина и тирозина в крови. Применяют тест Гатри, пробу Феллинга, флуориметрию, хроматографию, МРТ, поиск мутантного гена, электроэнцефалографию.
ЭЭГ позволяет обнаружить нарушения в основном в виде паттерна гипсартимии, даже если приступов у ребенка не наблюдалось. Также находят фокусы спайк- и полиспайк-разрядов (единичные и множественные). МРТ не находит изменений сигнала в стволе, мозжечке или коре головного мозга. Изменения на МРТ не коррелируют с уровнем интеллекта, они зависят от содержания фенилаланина в крови.
Если у ребенка фенилкетонурия II, то симптомы проявляются после 12 месяцев жизни. В крови затем находят повышенный уровень фенилаланина в периоде новорожденности, назначают диету, но болезнь всё равно прогрессирует. У малышей выраженная умственная отсталость, судороги, признаки повышенной возбудимости, гиперрефлексия, мышечная дистония, спастический тетрапарез. Летальный исход в части случаев наступает в возрасте от 2 до 3 лет.
Симптомы фенилкетонурии III напоминают выше перечисленные. У ребенка врачи обнаруживают три типичных признака:
- спастический тетрапарез
- микроцефалия
- глубокая умственная отсталость
Лечение Фенилкетонурии (ФКУ) у детей:
Новые методы лечения
На сегодня исследователи разрабатывают несколько методов альтернативной терапии фенилкетонурии (ФКУ):
- энзимотерапия фенилаланингидроксилазой, фенилаланинаммониалиазой
- метод «больших нейтральных аминокислот»
- лечение тетрагидробиоптерином (Сапроптерин)
Существует информация о случаях, когда пациентам с умеренной или легкой формой заболевания помогал тетрагидробиоптерин в дозе от 10 до 20 мг на 1 кг тела в сутки. В 2008 году было доказано, что для нормального физического развития детей с фенилкетонурией можно применять пищевые гликомакропептиды, которые также снижают содержание в плазме крови и головном мозге фенилаланина. Экспериментальным методом считается введение непосредственно в пораженные клетки печени ребенка введение гена фенилаланингидроксилазы. Этот метод пока не актуален в странах СНГ, в том числе Украине и России.
Продукты для детей с ФКУ (фенилкетонурией)
Диетотерапия позволяет предотвратить интеллектуальный дефицит при классической форме рассматриваемой болезни. Важное значение имеет возраст малыша, когда начинают применять диету. Каждый месяц без применения диеты малыш с ФКУ теряет примерно 4 балла IQ. Вопрос о диете при фенилкетонурии у детей в различных странах рассматривают по-разному. Но принципы едины.
При уровне уровень фенилаланина в крови до 2–6 мг% (120–360 мкмоль/л) у младенцев диета не применяется. Суть питания детей с ФКУ – в продуктах с низким содержанием фенилаланина, в основном это небелковая пища. Она нужна детям первого года жизни. В более позднем возрасте такое питание приносит меньше результатов.
Лечебный рацион питания при ФКУ:
- натуральные продукты питания
- лечебные продукты
- малобелковые продукты на основе крахмала
Детям с фенилкетонурией нельзя:
Смеси детям с ФКУ нужны только с минимальным содержанием белка. В течение первого года жизни допустимое количество фенилаланина составляет от 90 до 35 мг/кг ребенка. 50 мг фенилаланина = 1 г белка.
Лечебные продукты при фенилкетонурии у детей:
- ХР Аналог LCP
- MD мил ФКУ-0
- Афенилак
Диеты ребенку нужно придерживаться, если показатель фенилаланина в крови составляет минимум 360–480 ммоль/л.
Прикорм при фенилкетонурии (ФКУ у детей)
По достижению ребенком 3-месячного возраста рацион нужно расширять, вводя фруктовые и ягодные соки. Сначала это 3-5 капель, потом объем увеличивают до 30–50 мл. Для детей 12 месяцев доза уже составляет до 100 мл.
Соки в качестве прикорма:
- грушевый
- яблочный
- сливовый
Также в рацион постепенно вводят фруктовое пюре, постепенно увеличивая порцию. Детям от 4-4,5 месяцев уже можно овощное пюре, которое готовится родителями. Также можно плодовоовощные консервы для грудничков, но без молока. Второй прикорм – каша (10%) из безбелковой крупки или саго. Также ребенку можно давать безмолочные каши промышленного производства из кукурузной и/или рисовой муки. В них должно быть меньше 1 грамма белка на 100 мл готового продукта.
Детям от 6 месяцев можно вводить в диету кисели и/или муссы, в которых нет белка. Их готовят на амилопектиновом набухающем крахмале и фруктовом соке; это низкобелковый молочный напиток PKU «Лопрофин» и безбелковый напиток с молочным вкусом Нутриген. Детям с ФКУ от 7 месяцев можно давать низкобелковые изделия «Лопрофин»: рис, спагетти, спиральки. С 8 месяцев при фенилкетонурии малышам можно давать специальный безбелковый хлеб.
Диета для ФКУ у детей от 1 года
Для питания таких пациентов применяют продукты на основе смесей аминокислот без содержания фенилаланина и/или гидролизатов белка или с мизерным его количеством. В их составле должны быть комплексы макро-, микроэлементов и витаминов. По мере взросления ребенка дозу белка можно увеличивать, но не сразу. Количество углеводов и жиров нужно постепенно снижать, а потом и вовсе исключить. Рацион постепенно расширяется за счет натуральных продуктов и блюда.
Для детей с ФКУ от 12 месяцев можно применять специализированные лечебные продукты:
- Тетрафен 40
- Тетрафен 30
- MD мил ФКУ-1
- Тетрафен 70
- MD мил ФКУ-3
- Изифен
- П-АМ 1, П-АМ 2, П-АМ 3
- ХР Максамум (вкус нейтральный или апельсиновый)
- ХР Максамейд
Врачи советуют постепенно переходить на продукты для детей более старшего возраста на протяжении 1-2 недель. Объем предыдущей смеси нужно уменьшить на 1/4–1/5 и добавить эквивалентное по белку количество нового продукта. В части лечебных продуктов содержатся полиненасыщенные жирные кислоты. Среди малобелковых продуктов зарубежного производства есть напитки без белка, десерты, соусы и приправы, печенье и специальные сорта хлеба, которые можно детям при фенилкетонурии (ФКУ).
Часть исследователей склоняется к мнению, что детям с ФКУ диету нужно обогащать тирозином. Лечебные продукты имеют специфический вкус, потому могут быть нужны вкусовые добавки без содержания белка. Нельзя применять подсластитель аспартам, потому что при расщеплении он образует в том числе фенилаланин.
При лечении нужен регулярный контроль содержания фенилаланина в крови. Детей до 3 месяцев проверяют 1 раз в неделю, а после получения стабильных результатов – минимум 1 раз в 2 недели. Детей с ФКУ от 3 мес. до 1 года проверяют 1 раз в месяц, иногда – 2 раза. Для детей от 1 до 3 лет осмотры нужны минимум 1 раз в два месяца, а после трех лет контроль проводят 1 раз в 3 месяца.
Необходим контроль таких показателей для детей с ФКУ:
- физическое и интеллектуальное развитие ребенка
- нутритивный статус больного
- эмоциональное развитие
- речевое развитие
Один раз в месяц нужно проводить общий анализ крови. А по показаниям – биохимический анализ крови.
Если у ребенка обнаруживают дополнительные болезни с диспепсическими явлениями, интоксикацией, гипертермией, то диету можно прекратить на 2-3 дня, заменив лечебные продукты на натуральные, в которых не слишком много белка. Когда острая фаза болезни заканчивается, в рацион снова вводят лечебные продукты, но быстрее, чем в начале ввода диеты.
Прекращение диетотерапии
Возраст прекращения специальной диеты при ФКУ у детей до сих пор в процессе дискуссии. Существует информация, что при отмене диетотерапии в 5-летнем возрасте у одной трети детей с ФКУ отмечалось снижение уровня IQ на 10 баллов и более на протяжении следующих 5 лет. Прекращения диеты для детей старше 15 лет в некоторых случаях сказывались на прогрессирующих изменениях белого вещества мозга, что было выявлено при помощи МРТ.
При классической фенилкетонурии у детей диеты нужно придерживаться всю жизнь. Общее количество белка после наступления совершеннолетия не должно быть больше, чем 0,8–1,0 г/кг в сутки.
Профилактика Фенилкетонурии (ФКУ) у детей:
Чтобы организовать раннюю диетотерапию и избежать тяжелых церебральных повреждений, нужно проводить массовые скрининги на фенилкетонурию в неонатальном периоде. Это позволяет также избежать нарушения функционирования печени ребенка. Чтобы оценить риск рождения ребенка с рассматриваемым диагнозом, нужно предварительное генетическое консультирование для пар, у которых уже есть ребенок с фенилкетонурией (ФКУ) или у которых есть родственники с такой болезнью.
Женщины с фенилкетонурией до момента зачатия должны строго придерживаться диеты и продолжать ее, пока будут беременными. Это позволит избежать нарушений развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
Дети с ФКУ должны наблюдаться участковым педиатром и психоневрологом.
К каким докторам следует обращаться если у Вас Фенилкетонурия (ФКУ) у детей:
Педиатр
Психоневролог
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Фенилкетонурии (ФКУ) у детей, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Eurolab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом. Клиника Eurolab открыта для Вас круглосуточно.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.
Если Вами ранее были выполнены какие-либо исследования, обязательно возьмите их результаты на консультацию к врачу. Если исследования выполнены не были, мы сделаем все необходимое в нашей клинике или у наших коллег в других клиниках.
У Вас ? Необходимо очень тщательно подходить к состоянию Вашего здоровья в целом. Люди уделяют недостаточно внимания симптомам заболеваний и не осознают, что эти болезни могут быть жизненно опасными. Есть много болезней, которые по началу никак не проявляют себя в нашем организме, но в итоге оказывается, что, к сожалению, их уже лечить слишком поздно. Каждое заболевание имеет свои определенные признаки, характерные внешние проявления – так называемые симптомы болезни. Определение симптомов – первый шаг в диагностике заболеваний в целом. Для этого просто необходимо по несколько раз в год проходить обследование у врача, чтобы не только предотвратить страшную болезнь, но и поддерживать здоровый дух в теле и организме в целом.
Если Вы хотите задать вопрос врачу – воспользуйтесь разделом онлайн консультации, возможно Вы найдете там ответы на свои вопросы и прочитаете советы по уходу за собой. Если Вас интересуют отзывы о клиниках и врачах – попробуйте найти нужную Вам информацию в разделе Вся медицина. Также зарегистрируйтесь на медицинском портале Eurolab, чтобы быть постоянно в курсе последних новостей и обновлений информации на сайте, которые будут автоматически высылаться Вам на почту.
Другие заболевания из группы Болезни ребенка (педиатрия):
Если Вас интересуют еще какие-нибудь виды болезней и группы заболеваний человека или у Вас есть какие-либо другие вопросы и предложения – напишите нам, мы обязательно постараемся Вам помочь.www.eurolab.ua
Фенилкетонурия: основные сведения
Что такое фенилкетонурия
Фенилкетонурия – довольно редкое заболевание. Согласно статистике, 1 и 10 000 появившихся на свет новорожденных не обладает способностью усваивать фенилаланин – одну из самых распространенных в рационе питания человека аминокислот. Коварство этой болезни заключается в том, что без проведения специального анализа крови выявить ее наличие не представляется возможным, а обычная пища превращается в натуральный яд, отравляющий день за днем, год за годом мозг маленького человека. ФКУ без лечения приводит к самым тяжелейшим последствиям, проявляющимся в постепенном развитии слабоумия, вплоть до олигофрении и идиотии. Однако не все так уж и безнадежно, терапия фенилкетонурии существует и заключается в соблюдении специальной лечебной диеты в течение всей жизни.
Симптомы ФКУ
Первые симптомы фенилкетонурии можно заметить у малыша уже в трехмесячном возрасте. Чаще всего медики и родители обращают внимание на:
- Отставание в детском развитии
- Эпилептические приступы
- Нарушения мышечной спастики
- Слишком маленький размер мозга
- Неприятный запах мочи и пота (ацетон)
- Недостаток пигмента (обычно больные отличаются светлым цветом кожи и волос, голубыми глазами).
Диагностика ФКУ не представляет особой сложности. Для ее проведения достаточно лишь капли крови, взятой из пятки малыша через пару дней после рождения. В настоящее время анализ крови на фенилкетонурию входит в список обязательных процедур скрининга всех новорожденных.
Лечебная диета
Так как большинство продуктов правильного питания обычного человека содержит в своем составе фенилаланин (мясо, рыба, яйца, злаковые, молоко и кисломолочные продукты), ребенку, которому поставлен диагноз фенилкетонурия, требуется обеспечить соблюдение специальной безбелковой диеты. Аминокислоты, необходимые организму для роста и регенерации тканей, должны поступать в виде искусственно производимых смесей. Придерживаться диеты необходимо не только до полового созревания, но зачастую и пожизненно.
Кроме этого больным необходимо производить постоянный контроль уровня фенилаланина в крови.
Профилактика фенилкетонурии
Так как ФКУ является врожденным заболеванием генетического характера, никаких мер профилактики этой болезни не существует. Если семейный анамнез позволяет предполагать возникновение фенилкетонурии у будущего ребенка, женщине уже во время беременности стоит перейти на специальную диету, чтобы исключить вероятность негативного влияния фенилаланина на развивающийся мозг растущего плода.
medaboutme.ru